Genetics of Chronic Lymphocytic Leukemia
Department of Internal Medicine III, University of Ulm, Albert Einstein Allee 23, 89081 Ulm, Germany
Keywords
• CLL • Genetics • TP53 • ATM • IGHV • FISH
In general, chronic lymphocytic leukemia (CLL) is easy to diagnose; a peripheral blood sample for blood count, blood smear, and flow cytometry are sufficient. Whereas morphology and the immunophenotype of CLL cells are quite homogenous, there is a marked heterogeneity in the clinical course. The majority of patients do not require extended therapy, and the disease impacts them minimally; a distinct subgroup of patients, however, experiences rapid progression and insufficient response to treatment. The Binet and Rai staging systems 1,2 are accepted prognostic factors in CLL but these systems are not able to differentiate between good and bad prognosis,3 especially in the early stage. Cytogenetics provides a better insight into the pathogenesis of the disease and helps to estimate a patient’s prognosis more accurately. The discovery of different genomic aberrations that are associated with the prognosis has been very meaningful.4 Furthermore, the mutation status of the variable segments of the immunglobulin heavy chain genes (IGHV) has become one of the most important prognostic factors in CLL.5,6
Classical cytogenetics includes chromosome banding analysis, which had been difficult to achieve in the mostly nondividing CLL cells. Only recently have dedicated stimulation techniques brought forward this method and made it suitable for CLL diagnostics. In contrast, fluorescence in situ hybridization is also applicable in nondividing cells and has, therefore, found its way into CLL routine diagnostics.7 Analysis of chromosomal aberrations led to the discovery of important genes involved in CLL tumorigenesis such as TP53 and ATM.8,9 A closer look at recurrent chromosomal changes is provided by more advanced methods, with higher resolution like comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) array analysis. Moreover, DNA sequencing techniques have changed over the years, and today there are high-throughput methods of particular sensitivity that are embraced by the term “next generation sequencing.”
Fluorescence In Situ Hybridization
In a comprehensive study, at least one chromosomal aberration was detected by FISH in 268 of 325 cases of CLL (82%).4 The most common aberrations were deletion in 13q (55%), deletion in 11q (18%), trisomy of 12q (16%), deletion in 17p (7%), and deletion in 6q (6%). Correlation of the cytogenetic data with clinical and laboratory findings led to a hierarchical model of 5 major categories with different prognostic outcome: 17p deletion, 11q deletion without 17p deletion, 12q trisomy without 17p deletion or 11q deletion, normal karyotype and 13q deletion as the sole abnormality (Fig. 1). Patients with 17p deletion have the worst prognosis. This deletion affects the tumor suppressor gene TP53 and is associated with refractoriness to conventional chemotherapy with fludarabine. The ataxia teleangiectasia mutated (ATM) gene is located on the long arm of chromosome 11. Its deletion has been shown to be associated with poorer outcome and marked lymphadenopathy.4 In a large randomized, open-label, phase 3 trial (CLL8 trial of the GCLLSG, NCT00281918) on FCR (fludarabine, cyclophosphamied, rituximab) as first-line treatment, the negative prognostic effect of the 17p deletion was not abrogated with chemoimmunotherapy, whereas the rate of complete remissions in 11q deleted cases was increased by more than times. This interesting result suggests that the bad prognostic significance of the 11q deletion might be overcome by this effective regimen and that a specific first-line treatment might change the natural course of the disease.10
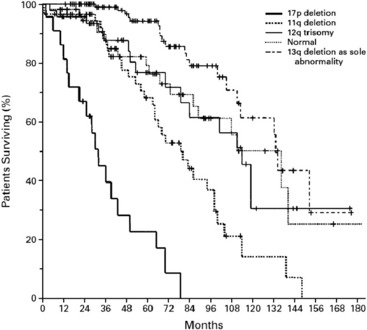
Fig. 1 The hierarchical model of chromosomal abnormalities in CLL: Probability of survival related to the time after diagnosis among the genetic subgroups.4
There is consensus to use interphase FISH in routine practice in CLL at the time of treatment, mainly to detect patients with a 17p deletion who will not respond to conventional chemotherapy and who will experience early progression and short survival after therapy.7,11 These patients might benefit from agents that act independently of p53 like alemtuzumab, flavopiridol, and allogeneic stem cell transplantation.12,13 Moreover, as the cytogenetic profile can change over the course of the disease,14 these diagnostics might be repeated before subsequent therapies.
TP53 Mutation
The deletion of 17p13 in CLL affects the tumor suppressor gene TP53. In 80–90% of the cases, a deletion of 17p is associated with a mutated TP53 gene on the remaining copy.15 This is considered one reason why p53 pathway-based therapies like fludarabine or its combinations such as fludarabine/cyclophosphamid (FC), fludarabine/cyclophosphamide/rituximab (FCR), etc, are not effective in patients with 17p deletion. TP53 mutations have been found in 4% to 15% of patients in early stage untreated CLL and are associated with a poorer outcome.15–17 Mutations of TP53 independently of a 17p deletion have been shown in 4.5% of the cases in a recent analysis of the randomized prospective CLL4 trial of the GCLLSG (randomized fludarabine vs fludarabine/cyclophosphamide [FC] as first-line treatment). Progression-free survival and overall survival of patients with a sole TP53 mutation is similar to those of patients with 17p deletion (Fig. 2).15
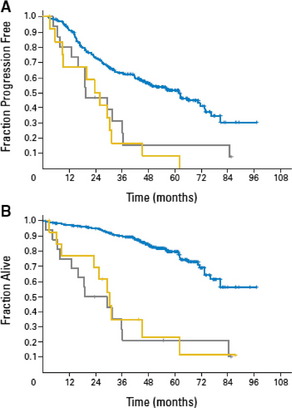
Fig. 2 Outcome of patients with sole TP53 mutation (yellow) and patients with 17p deletion (gray) compared to the remaining patients (blue)15: (A) The median progression free survival was significantly shorter for patients with sole TP53 mutation (23.3 months) or 17p deletion (19.2 months) compared to the remaining patients (61.8 months, P<.001). (B) The median overall survival was significantly shorter for patients with sole TP53 mutation (30.2 months) or 17p deletion (19.2 months) compared to the remaining patients (median OS not reached, P<.001).
ATM Mutation
The kinase ATM is activated by DNA double-strand breaks and targets P53, among others. The ATM gene is localized in the minimal consensus region in bands 11q22.3 to 11q23.1. Deletion of 11q has been shown in 18% of CLL patients.4 In about one third of 11q-deleted patients, a simultaneous mutation of ATM18 has been found. Overall survival is shorter in patients with an 11q deletion combined with an ATM mutation of the remaining allele compared to a sole 11q deletion (Fig. 3). Deletion of 11q as well as ATM mutations are associated with poorer outcome.4 Furthermore, an association between mutation of the ATM gene and unmutated IGHV genes has been shown.19
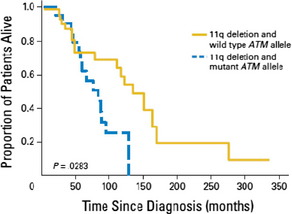
Fig. 3 Overall survival from diagnosis of patients with 11q deletion and wild type ATM allele compared to patients with 11q deletion and mutant ATM allele: The overall survival was significantly shorter for patients with 11q deletion and ATM mutation compared to patients with 11q deletion and wild type ATM allele (P = .0283).18
Chromosome Banding Analysis
Compared to FISH, chromosome banding analysis (CBA) provides an overview of the whole genome but is limited to dividing cells during metaphase. Chronic lymphocytic leukemia cells have a low mitotic activity and, therefore, this technique has not been applicable until recently.20
However, stimulation with CD40 ligand or the combination of CpG-oligonucleotides and IL-2 has achieved an increase in metaphase spreads.21 Balanced and unbalanced translocations were seen in 34% of the patients. Most of them had not been previously described and were not recurrent in the cohort. In a large study on 506 CLL samples, 98.8% were successfully stimulated by the immunostimulatory CpG-oligonucleotide DSP30 and IL-2. Chromosome banding analysis detected 83% aberrations compared to 78.4% found by FISH. In addition, a subgroup with a complex aberrant karyotype was seen, which was associated with an unmutated IGHV-status and expression of CD38.22 Chromosome banding analysis, therefore, detects additional abnormalities and might complement FISH-generated data. However, further studies are needed to determine the value of CBA for prognosis and treatment.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


