Fig. 1
Normal thyroid gland with immunohistochemically demonstrated C cells. The predominantly disperse distributed C cells are located intrafollicular (calcitonin immunohistochemistry, ×200)
Some decades later, electron microscopic studies supported the observation (DeLellis et al. 1978) that human C cells are indeed intrafollicular and separated from the thyroid interstitium by the follicular basement membrane. C cells contain moderately electron-dense larger (type I; 280 nm) and more electron-dense smaller (type II; 130 nm) secretory granules; ultrastructural immunohistochemistry demonstrated the peptide hormone calcitonin in both type I and type II granules. Besides, these granules C cells contain abundant endoplasmic reticulum and mitochondria. All these structures are also detectable in hyperplastic and neoplastic C cells.
2 Embryology of C Cells
The majority of C cells in the human thyroid gland are originating from primordial C cells of the neural crest, which migrate ventrally to the ultimobranchial body (a derivative of the ventral recess of the fourth pharyngeal pouch). These C cells are thus of neuroectodermal origin. During the descent of the thyroid, the ultimobranchial body is incorporated into both thyroid lobes, ideally leading to a disperse distribution of the C cells in preferred regions of the thyroid lobes (see below). Apparently in most humans, the ultimobranchial body is completely incorporated into the thyroid lobes. However, occasionally C cells remain along their migration path in extrathyroidal location and may give rise to pathological processes of C cells (hyperplasia and neoplasia) outside the thyroid (Hirsch et al. 2004; Smets et al. 1990). Disturbances in the intrathyroidal distribution result in focal accumulation of C cells (Gibson et al. 1980). Studies in thyroid glands of patients with DiGeorge syndrome confirmed on the one hand the origin of C cells from the ultimobranchial body (Pueblitz et al. 1993), and on the other hand, they suggest that a (small) subset of C cells most likely arise from endodermal stem cells (Harach 1997).
3 Normal Human C Cells
In the thyroid, three types of epithelial cells can be distinguished: follicular cells accounting for more than 99.9 % of all thyroid epithelia, C cells (0.01–0.1 %) and the only occasionally demonstrable so-called solid cell nests (SCN); the latter most likely represent remnants of the ultimobranchial body, which is supported by the calcitonin production of SCN. The number of C cells is higher in newborns than in adults and increases again after 60 years of age (O’Toole et al. 1985).
C cells are mainly confined to groups of thyroid lobules located centrally in the rear upper portions of both thyroid lobes. At the poles of the thyroid lobes, the isthmus and pyramidal lobe C cells are detectable in significantly reduced numbers or completely missing. Thus, medullary thyroid carcinoma (MTC) may occur in all parts of the thyroid; however, it can be found more frequently in areas of high C-cell concentration than in the peripheral thyroid.
Histologically, C cells are difficult to identify on routine H&E sections; usually, calcitonin immunohistochemistry is performed to demonstrate C cells on the light microscopical level (LiVolsi 1990). The majority of C cells (size up to 40 µm) are round or polygonal, found singly or in groups of 3–5 cells. A second type of C cells is spindled shaped with tapered ends (Ljungberg 1972; Schmid et al. 1992). Virtually, all C cells are situated within the follicular basement membrane.
4 Function of C Cells
The term “C cell” was introduced by Copp and Cameron in 1961, who postulated in isolated thyroid and parathyroid glands of dogs a type of cell producing and secreting a hormone “calcitonin” with calcium-lowering effect (Copp and Cameron 1961); however, initially, the parathyroid glands were assumed as the origin of the hormone (Copp et al. 1962). In 1964, it was recognized that calcitonin originates from the thyroid gland (Foster et al. 1964); in 1967 (Tauber 1967), the previously postulated “C cells” were morphologically demonstrated in the thyroid gland and their neuroectodermal origin was revealed. Already in 1966, E. Dillwyn Williams had predicted the until then postulated C cells as the source of MTC (Williams 1966). In 1973, Hubert J. Wolfe recognized that C-cell hyperplasia (CCH) is an obligatory precursor of hereditary MTC (Wolfe et al. 1973).
Almost all human C cells synthesize and secrete calcitonin; only in a small fraction of the C cells exclusively calcitonin gene-related peptide (CGRP) is detectable. Calcitonin is a 32-amino acid linear polypeptide hormone. Structurally, calcitonin has an intramolecular disulphide bridge (Cys-1 to Cys-7) and an amidated C-terminus; both are essential for the biological activity of the hormone. Disulphide bridge-free calcitonin is able to bind specifically to the calcitonin receptor; however, it acts at the receptor as a competitive antagonist. The neuropeptide CGRP consists of 37 amino acids and is encoded by the same gene as calcitonin; it occurs predominantly in the peripheral and central nervous system and to a lesser extent in the thyroid by selective mRNA splicing.
An increase of serum calcium in the blood, gastrointestinal hormones and pentagastrin (a synthetic oligopeptide with gastrin effect) stimulate calcitonin secretion. To lower the serum calcium level, calcitonin induces a decreased osteoclastic activity in the bone, an increased calcium excretion from the kidney and a reduced resorption of calcium in the intestine. However, the efficacy of calcitonin in the regulation of calcium in comparison with the other calcium-regulating hormones calcitriol and parathyroid hormone is relatively limited.
5 C-Cell Hyperplasia (CCH)
An increase in size and number of C cells is called CCH. The term CCH refers, however, to two different C-cell conditions with completely different pathological potential (Albores-Saavedra and Krueger 2001; LiVolsi 1997; Perry et al. 1996). Inherited “neoplastic C-cell hyperplasia”, by definition always associated with germline mutation of the RET proto-oncogene, is the precursor lesion of familial MTC and can easily be identified on H&E sections (DeLellis et al. 2004). The enlarged, pale C cells are mainly located in areas of high C-cell concentration (Ting et al. 2015). Neoplastic CCH is found in prophylactic thyroidectomy specimens of asymptomatic carriers of the RET-mutation or patients with MTC usually in close vicinity of (multifocal) invasive familial MTC. Morphologically, neoplastic CCH is characterized by groups of atypical cells situated within the thyroid follicular basement membrane (focal CCH); these cells may encircle the entire follicular lumen (diffuse CCH; Fig. 2) and ultimately obliterate the follicular lumen partially or completely (nodular CCH; Fig. 3). Neoplastic CCH may be difficult to distinguish from SCN, palpation thyroiditis and occasionally papillary microcarcinoma (Elisei et al. 2008; Schmid and Sheu 2015).
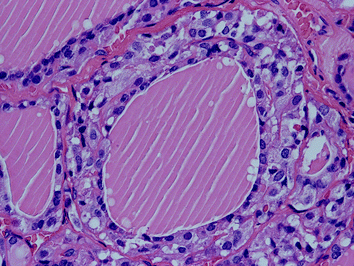
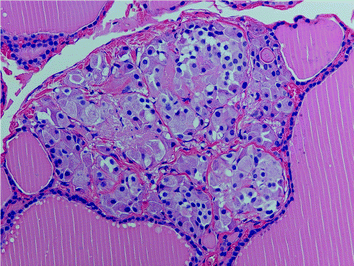
Fig. 2
“Diffuse” neoplastic C-cell hyperplasia (MEN 2A) with atypical cells encircling the entire follicular lumen (H&E ×200)
Fig. 3
“Nodular” neoplastic C-cell hyperplasia (MEN 2A) with complete obliteration of the follicle by atypical C cells. The follicular basement membrane is still intact (H&E ×200)
In contrast, “non-MEN2-associated CCH” (Ting et al. 2015), previously named “sporadic” or “physiological” CCH (Perry et al. 1996), cannot be identified on H&E sections (DeLellis et al. 2004); its identification requires immunohistochemical calcitonin demonstration (Fig. 4). Non-MEN2-associated CCH is defined as an increase of normally appearing C cells; at 100-fold magnification, at least 50 C cells (calcitonin immunohistochemistry) have to be identified in areas of high C-cell concentration (Rosai et al. 1992). Occasionally, non-MEN2-associated CCH can also show focal obliteration of follicles. Non-MEN2-associated CCH has been described in association with a great variety of pathological conditions, including non-medullary thyroid tumours (Albores-Saavedra et al. 1988; Scheuba et al. 2000), autoimmune thyroiditis Hashimoto and thyroid non-Hodgkin-lymphoma (Baschieri et al. 1989), thyrotoxicosis (Scheuba et al. 2000), in the vicinity of SCN (Chan and Tse 1989), renal failure and calcium metabolism disorders. Up to 50 % of patients with nodular goitre (age- and sex-independent) show a morphologically detectable non-MEN2-associated CCH without increased serum calcitonin (Kaserer et al. 1998). To the best of knowledge, this form of CCH is not related to the development of sporadic MTC; so far, no somatic mutations of the RET proto-oncogene could be demonstrated in the C cells of “non-MEN2-associated” CCH (Saggiorato et al. 2007).
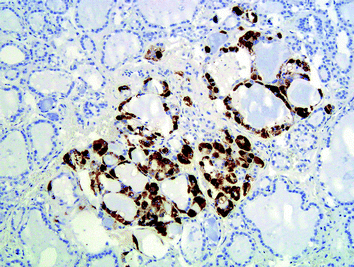
Fig. 4
“Non-MEN2-associated” C-cell hyperplasia, which cannot be demonstrated on H&E sections; the increased number (>50 at 100-fold magnification in areas of high C-cell concentration) of normally appearing C cells is demonstrated by immunohistochemistry (calcitonin antibodies; ×100)
6 Transition of Neoplastic CCH to Familial MTC
The differential diagnosis of neoplastic CCH from microinvasive familial MTC may cause severe difficulties (DeLellis et al. 2004). Microinvasive MTC is characterized by defects of the follicular basement membrane (Rosai et al. 1992) which are virtually impossible to demonstrate on routine H&E sections. However, this is regularly accompanied by the development of stromal desmoplasia around the tumour cell nests which is a useful and reliable surrogate marker of tumour cell invasion (Sheu and Schmid 2010; Fig. 5). In some cases, the neoplastic C-cell aggregates are measuring several millimetres apparently without developing stromal desmoplasia (Fig. 6a, b); due to their size, these lesions are classified as MTC although no generally accepted consensus on this issue exists.
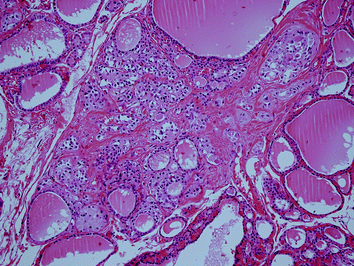
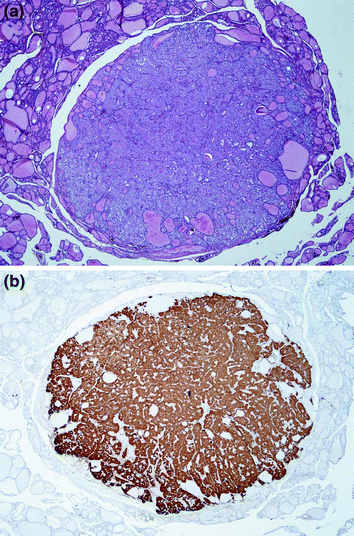
Fig. 5
Invasive familial MTC (MEN 2A) with stromal desmoplasia and tumour cell invasion. The MTC is measuring approx. 3 mm (H&E ×100)
Fig. 6
a Familial MTC (MEN 2A) without stromal demoplasia or obvious invasive growth in the tumour periphery; the diagnosis of MTC is based on its size (5 mm). (H&E ×50). b Immunohistochemical calcitonin demonstration in the MTC depicted in Fig. 6a (×50)
7 Medullary Thyroid Carcinoma (MTC)
Medullary carcinoma (MTC; ICD-O 8345/3) is defined as a malignant tumour of the thyroid gland showing C-cell differentiation (DeLellis et al. 2004). Up to 25 % of MTC is caused by a gain of function mutation of the RET proto-oncogene (familial MTC); familial MTC is preceded by neoplastic CCH hyperplasia. Sporadic (non-inherited) MTC has no defined precursor lesion; 40–60 % of sporadic MTC shows somatic mutations of the RET proto-oncogene (Elisei et al. 2008; Hofstra et al. 1994). The vast majority of MTC synthesizes and secretes the peptide hormone calcitonin. Thus, calcitonin is a very useful immunohistochemical markers for the histological diagnosis of MTC (Harach et al. 1992; Schmid and Böcker 1993). Rarely, however, MTC may be lacking immunohistochemically detectable calcitonin (“atypical MTC” (Schmid and Ensinger 1998)), and/or patients suffering from histologically proven MTC may lack pathologically elevated serum calcitonin (“non-secretory MTC” (Frank-Raue et al. 2013)).
In 1951, Robert C. Horn described (Horn 1951) a series of seven cases of a variant of thyroid carcinoma with solid growth pattern and amyloid, which he classified subsequently as poorly differentiated and highly malignant (Horn and Dull 1951). The term “MTC” was coined in 1959 by Hazard et al. based on a series of 21 thyroid carcinomas, again morphologically appearing as solid tumours with amyloid deposits; biologically, the tumours were considered to have an intermediate position between differentiated (papillary and follicular thyroid carcinoma) and anaplastic thyroid carcinomas (Hazard et al. 1959). In 1966, E. Dillwyn Williams recognized striking similarities between human medullary carcinoma and thyroid tumours in dog and rat originating from “parafollicular cells” (Williams 1966). He proposed that human medullary carcinoma might be derived from the to that time in men still postulated C cells, and he predicted that if the C cells were the source of calcitonin, medullary carcinoma might also produce this hormone. In 1968, Meyer and Abdel-Bari demonstrated electronmicroscopically secretory granules in the tumour cells as well as biochemically a 100-fold increased calcitonin concentration in the tumour compared to normal thyroid tissue (Meyer and Abdel-Bari 1968). Using immunofluorescence, Bussolati et al. demonstrated in 1969 calcitonin in the cells of medullary(Bussolati et al. 1969).
In 1886, Felix Fränkel reported of an already 1884 performed autopsy of a 18-year-old female patient with bilateral adrenal tumours (then interpreted as an angiosarcoma) and goitre (Fränkel 1886); this is considered to be the first description of a case of multiple endocrine neoplasias type 2A. Over 120 years later, a germline mutation in the RET proto-oncogene at codon 634 TCG > TGG (C634W) was demonstrated on the still available tissue specimens of the patient; subsequently, descendants of brothers of the patient suffering from pheochromocytomas and MTC were identified (Neumann et al. 2007). John H. Sipple described in 1961 the combination of pheochromocytoma with adenocarcinoma of the thyroid (Sipple 1961); subsequently, the combination was recognized as the (autosomal-dominant inherited) syndrome of multiple neuroendocrine neoplasia (MEN) 2A (Steiner et al. 1968), in which the in all cases occurring MTC determines largely the patients’ survival (Machens and Dralle 2006). The first description of a case with MEN 2B was made in 1922 by Wagenmann (1922) and Froebius (1922).
Up to date, no benign C-cell tumour has been defined. In familial MTC, malignancy is presumed when hyperplastic C cells show invasive growth through the follicular membrane which is accompanied by the development of a fibrous desmoplastic stroma (Rosai et al. 1992; Sheu and Schmid 2010; Fig. 5). This stromal desmoplasia is also observed in approximately 80 % of sporadic MTC. However, in the remaining 20 % of sporadic MTC as well as in a number of familial MTC, stromal desmoplasia is completely lacking (Koperek et al. 2008); in these cases, which are usually well circumscribed but not necessarily enveloped by a tumour capsule, the diagnosis of MTC (by definition a malignant tumour! (DeLellis et al. 2004)) is apparently rather based on the size of the C-cell tumour than the demonstration of unequivocal invasive growth (Synoracki et al. 2015; Fig. 6a, b).
7.1 Morphology of MTC
MTC is mainly located in the lateral upper two-thirds of the thyroid lobes, the area of highest C-cell concentration; the tumours range in size from barely visible to several centimetres in diameter (LiVolsi 1990). In >90 %, familial MTC is associated with (small) multifocal and bilateral tumours, whereas in sporadic MTC, multifocality is the exception (Rosai et al. 1992; Schmid et al. 2003). In the isthmic region or the thyroid periphery, MTC is located only occasionally. Macroscopically, MTC presents with a greyish-white to reddish-brown cutting surface; the tumours are often circumscribed and in rare cases even completely encapsulated. On the other hand, occasionally even very small tumours (<7 mm) may macroscopically already show infiltrative borders (Chan 2007). With increasing size, the tumours may regularly develop haemorrhage, cystic regressions and/or in large tumours central necrosis (DeLellis et al. 2004).
The histopathological appearance of MTC is exceptionally variable (DeLellis et al. 2004; LiVolsi 1990; Rosai et al. 1992; Synoracki et al. 2015). MTC may mimic a broad variety of thyroid and non-thyroid tumours; thus, every unusual thyroid tumour should be investigated using calcitonin immunohistochemistry in order to prove or exclude MTC (Schmid et al. 2003). The typical MTC shows a solid and compact growth pattern of nests of polygonal- and spindle-shaped tumour cells which regularly freely infiltrate into the surrounding non-neoplastic thyroid tissue. The cytoplasm of the tumour may appear granulated, and the predominantly uniform round-to-oval nuclei show a coarsely granulated chromatin (“salt-and-pepper pattern”). The number of mitosis found is quite variable. The tumour stroma may show delicate collagen bands without stromal desmoplasia; MTC without stromal desmoplasia is statistically significantly associated with a very low potential for metastasis (Koperek et al. 2008; Scheuba et al. 2006). So far, no specific immunohistochemical and/or molecular markers have been found further characterize MTC without stromal desmoplasia (Koperek et al. 2007, 2009, 2011). In the majority of MTC (approx. 80 %), stromal desmoplasia, indicating invasive growth of tumour cells, can be demonstrated; however, the extent of stromal desmoplasia may vary considerable.
Amyloid deposits are present in 60–85 % of MTC cases. The demonstration of amyloid may be strongly suggestive for the diagnosis of MTC; however, since amyloid can be found in a variety of non-C-cell differentiated thyroid lesions, the diagnosis of MTC cannot be based exclusively on the demonstration of amyloid.
7.2 Histological Variants
The papillary or pseudopapillary variant of MTC is characterized by the presence of true papillae or artificial pseudopapillary structures; the tumour cell nuclei are lacking the nuclear features of papillary carcinoma (Fig. 7). MTC with glandular features (follicular or trabecular) may cause differential diagnostic problems with adenomas and follicular carcinomas (Fig. 8). The insular variant of MTC may resemble poorly differentiated thyroid carcinoma or classical carcinoid. Giant cell and spindle cell (Fig. 9) variants of MTC may be confused with anaplastic thyroid carcinoma; although these variants of MTC may show a less favourable outcome, the prognosis is still much better than that of anaplastic carcinoma. Small-cell variant of MTC, which is more commonly associated with tumour necrosis than other variants of MTC, is also associated with a poorer prognosis. The oncocytic variant of MTC may be misdiagnosed as oncocytic (Hürthle cell) variants of other thyroid tumour entities; since oncocytic thyroid tumours with follicular cell differentiation may show a false-positive calcitonin immunoreactivity (due to the antibody clone used), the diagnosis of the oncocytic variant of MTC should be confirmed by additional immunohistochmical markers [e.g. chromogranins, synaptophysin, carcinoembryonic antigen (CEA)]. The paraganglioma-like variant of MTC (Fig. 10) contains numerous S-100-positive sustentacular cells; in contrast to this MTC variant, the rare primary intrathyroidal paraganglioma is negative for cytokeratins. The angiosarcomatoid variant of MTC (Fig. 11) is characterized by a variable number of vascular spaces containing erythrocytes. Rarer variants of MTC include the clear cell, squamous cell, melanin-producing and amphicrine variants. However, it has to be emphasized that the morphological variants of MTC rather highlight the danger of a misdiagnosis then represent “real” entities with a defined biological behaviour.
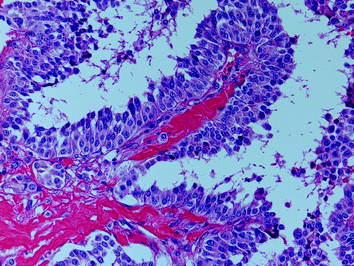
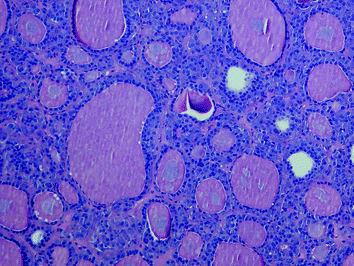
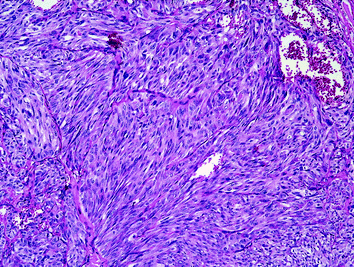
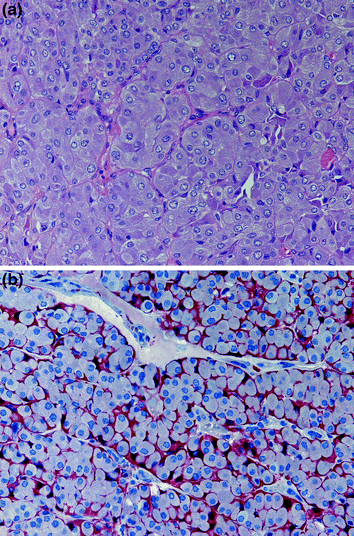
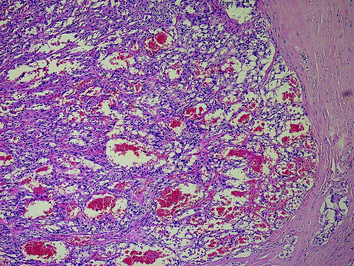
Fig. 7
Papillary structures in a sporadic MTC; the tumour cell nuclei show a coarsely granulated chromatin (“salt-and-pepper pattern”) without the morphological nuclear features of papillary thyroid carcinoma (H&E ×200)
Fig. 8
Glandular (follicular) features in a sporadic MTC; this growth pattern may cause differential diagnostic problems with follicular neoplasms (H&E ×100)
Fig. 9
Spindle cell variant of MTC may be confused with anaplastic thyroid carcinomas (H&E ×200)
Fig. 10
a Paraganglioma-like variant of MTC (H&E ×200). b Numerous S-100-protein positive sustentacular cells in a paraganglioma-like variant of MTC (S-100-protein immunohistochemistry, ×200)
Fig. 11
Angiosarcomatoid variant of MTC with prominent vascular spaces containing erythrocytes (H&E ×100)
7.3 Immunohistochemistry
Calcitonin is expressed, although in considerable different quantity, in the vast majority of MTC (DeLellis et al. 2004; Harach et al. 1992; Krisch et al. 1985; Lloyd et al. 1983; Schmid and Ensinger 1998); by combined use of calcitonin and chromogranin A antibodies virtually, all MTC can be immunohistochemically detected (Harach et al. 1992). MTC expresses general neuroendocrine markers such as chromogranins (Schmid et al. 1987), synaptophysin and others (Chan 2007). Additionally, a broad variety of peptides (somatostatin, ACTH, serotonin, gastrin, bombesin, calcitonin gene-related peptide (CGRP) and others) can be demonstrated (Chan 2007; Schmid and Böcker 1993). CEA is expressed in most cases; in less differentiated MTC, CEA is usually stronger expressed than calcitonin (Lloyd et al. 1983; Schmid et al. 2003; Schröder and Klöppel 1987). Nuclear TTF-1 and Islet-1 expression can be found in most MTC (Agaimy et al. 2013). S-100 positive sustentacular cells can be regularly found in MTC (more often in familial than in sporadic MTC (Matias-Guiu et al. 1998)). Numerous S-100 positive sustentacular cells are the immunohistochemical hallmark of the paraganglioma-like variant of (sporadic) MTC (Bockhorn et al. 2005). Rarely, MTC may (almost completely) lack calcitonin and CEA expression (Bockhorn et al. 2004; Krisch et al. 1985); these tumours are referred to as “atypical MTC” (Schmid and Ensinger 1998). Even rarer patients with histologically proven MTC may lack pathological elevation of serum calcitonin (“non-secretory MTC” (Frank-Raue et al. 2013)); in these cases, MTC may immunohistochemically either show very few individual and weekly calcitonin-positive cells (as in “atypical MTC”), and in other cases, however, an unequivocal stronger calcitonin immunoreactivity can be demonstrated in the majority of tumour cells (Frank-Raue et al. 2013). Completely, calcitonin-negative MTC may strongly express CGRP (Nakazawa et al. 2014). Calcitonin-negative tumours of the thyroid with neuroendocrine features (immunohistochemistry) may represent metastases from other neuroendocrine carcinoma (predominatly from lung or gastrointestinal tract), an intrathyreoidal located parathyroid tumour or rare primary paraganglioma of the thyroid.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


