Introduction
The global prevalence of pediatric obesity has increased by a staggering eightfold over the past 4 decades with a current estimate of 124 million children, ages 5 to 19, having obesity worldwide. The concept of the adipocyte functioning as an endocrine organ rather than simply serving as a repository for lipid storage emerged a quarter of a century ago when leptin was identified as the first of many adipokines produced by fat tissue. Thus given that the pathophysiology of obesity is a form of endocrine derangement at its core, and because many of the complications associated with obesity have an endocrine component in nature, pediatric endocrinologists are increasingly receiving referrals for the treatment of obesity and its comorbidities. Type 2 diabetes, which was once rare in children and is typically associated with obesity, now comprises approximately one-third of all new diagnoses of pediatric diabetes and continues to rise by nearly 5% in annual incidence rate among youth.
This chapter provides a framework for understanding the regulation of energy balance and summarizes approaches for assessing and managing pediatric patients with obesity. On the flip side of the energy-balance coin, disorders of energy inadequacy are also discussed in this chapter, and they provide insight on the converse problem of nutritional insufficiency. Although the relative dearth of current knowledge on how best to ameliorate the complex biologic, behavioral, and environmental contributors to energy imbalance poses a therapeutic challenge, recent advances in novel “precision medicine” approaches that target specific defects in energy homeostasis provide insights into potentially effective treatments for both obesity and underweight disorders.
Energy balance
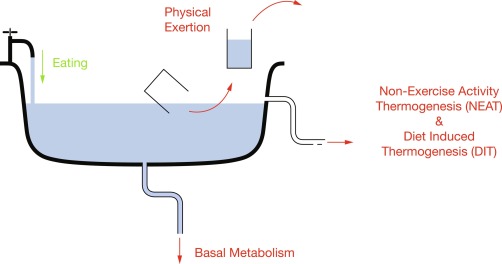
The major components of energy balance are energy intake, energy expenditure, and energy storage. Sources of energy intake include carbohydrate (4 kCal/g), protein (~ 4 kCal/g), fat (9 kCal/g), and alcohol (7 kCal/g), typically negligible in children, but possibly significant in some adolescents. Energy expenditure is comprised of basal metabolic rate ([BMR], energy needed to maintain bodily functions, highly correlated with lean muscle and organ mass, and affected by various disease states), thermic effect of food (energy used to digest food, diet-induced thermogenesis [DIT]), and physical activity (exercise and nonexercise activity thermogenesis [NEAT], which includes activities of daily living, fidgeting, and posture maintenance). Energy storage, primarily in the form of fat, occurs when intake exceeds expenditure. Using a bathtub ( Fig. 24.1 ) as an analogy for energy balance with the water level representing total energy stores, the water volume (total body fat) is determined by intermittent addition of water from the faucet (meals), episodic removal of water buckets (physical exertion), continual drainage of water from a bottom drain (BMR), and overflow drainage of water from an upper drain (NEAT and DIT) that is positioned at the set point for maximal water level (individual homeostatic set point). Changes in faucet flow rate, size of buckets, aperture diameter of the drains, and height of overflow set point together determine the water level in the bathtub.
Human energy balance is regulated by intricately connected homeostatic and non-homeostatic mechanisms. The homeostatic system maintains body fat stores within a fairly tight range for each individual, remarkably within around 10 kCal/d on average, such that even small perturbations in balance can lead to cumulative weight gain over time. The non-homeostatic system mediates environmental and cognitive factors that motivate eating, including the reward aspects of food intake and learned behaviors that lead to food consumption for reasons other than fulfillment of nutritive needs. The neural circuitry and neuroendocrine hormones of the homeostatic and non-homeostatic processes interact and overlap to form a complex system that integrates temporal, spatial, and contextual signals of metabolic status and higher-order brain function. Because survival depends on an adequate supply of energy, the evolutionary calibration of energy balance favors food consumption and promotes excess energy storage to protect against starvation when food is scarce. Consequently, in our modern era, in places where food is readily available in abundance, especially highly palatable, energy-dense foods, the inhibitory mechanisms for appetite regulation may be inadequate for preventing overconsumption.
Regulation of Food Intake
Human meal patterning is typically characterized by discrete bouts of food consumption interspersed with periods of fasting. The time-course of each meal follows a cycle, comprised of cephalic, gastric, and intestinal phases followed by a postabsorptive state. The cephalic phase consists of the physiologic responses to the thought, sight, smell, and taste of food in anticipation of ingestion. In the gastric phase, ingested food enters the stomach and digestion formally begins. As food leaves the stomach, the intestinal phase begins, where secreted pancreatic enzymes enhance digestion and nutrient absorption. Finally, in the postabsorptive state, nutrient absorption from the meal is complete and circulating glucose concentrations are maintained by glycogenolysis and gluconeogenesis until initiation of the next meal.
Short-term regulation of appetite determines satiety (time period between meals before hunger prompts meal initiation) and satiation (sensation of having eaten enough, leading to meal termination). Food intake is driven by a combination of hunger, social context, and sensory inputs. Hunger is mediated by a drop in anorexic (appetite-suppressing) signals in the postabsorptive state following the prior meal, and by a rise in ghrelin, an orexigenic (appetite-stimulating) peptide secreted by the enteroendocrine cells of the stomach during fasting. Nutrient ingestion leads to a rise in anorexic signals, including gastric wall stretch and mechanical contact with food, secretion of intestinal peptides (e.g., cholecystokinin [CCK], peptide YY, glucagon-like peptide 1 [GLP-1], etc.), and entry of digested nutrients into circulation. The vagal nerve is the primary neural connection between the gastrointestinal (GI) tract and the central nervous system (CNS). It is the longest cranial nerve and contains both sensory and motor fibers involved in the regulation of parasympathetic (“rest and digest”) homeostasis. Vagal sensory afferents from the gut terminate in the hindbrain where connections to cortical, forebrain, and midbrain regions are involved in vagal motor efferent regulation of GI motility and secretory functions for digestion. Importantly, the GI tract is also innervated by the spinal nerves, which convey sensory inputs from the intestinal tract to the homeostatic centers. Satiation is reached when ghrelin drops; gastric distension triggers anorexic vagal afferents to the hindbrain; and increases in glucose, insulin, and anorexic peptides lead to slowing of gastric motility and signaling of fullness to the CNS. Satiety, which determines the interval until the next meal, is influenced by the quantity and composition of the prior meal and additional physiologic contributors, such as gut microbiota, fermentation products, and bile acids.
Nonnutritive aspects of meal regulation include food palatability and neuropsychologic factors, such as mindfulness, stress, cognitive demands, and alertness. Food craving is linked to the hedonic aspects of eating because of activation of the mesocorticolimbic reward pathway. Opioid receptor activation mediates the reward sensation of food and the pleasure of eating, whereas dopaminergic activation mediates the reward value of food and the motivation to obtain food. Social context (gatherings and events where food consumption is expected) and sensory inputs (sight and smell of palatable food) can drive food intake in the absence of hunger, making environmental contingencies and stimulus control just as critical to address as physiologic cues for obesity management.
Homeostatic system for energy balance
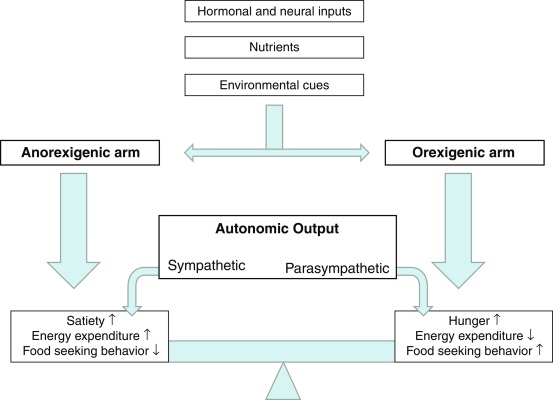
The CNS is the control center for energy homeostasis. Afferent signals from the periphery deliver energy status information to the brain, which processes these cues and then delivers efferent signals to modulate energy intake and expenditure. The system is directed by several hypothalamic nuclei, which receive an array of hormonal, neural, environmental, and cortical inputs. These signals activate, based on their directionality, anorexigenic or orexigenic signal transduction pathways. The output of the system is conveyed via the autonomic nervous system and is translated by peripheral organs into food-seeking behavior, energy conservation/wasting, and physiologic adaptations (a simplified vision of these pathways is shown in Fig. 24.2 ). The physiologic target of energy homeostasis is to maintain stable levels of total body fat rather than overall weight per se. Therefore leptin, which is secreted in proportion to total fat mass, functions suitably as a key circulating signal to convey adiposity status to the CNS. Binding of these hormones to their respective receptors within the hypothalamic arcuate nucleus (ARC) upregulates anorexic/catabolic signals from proopiomelanocortin (POMC)/cocaine-amphetamine–related transcript (CART) expressing neurons and downregulates orexigenic/anabolic signals from Agouti-related peptide (AgRP)/neuropeptide Y (NPY) expressing neurons. A schematic of these energy homeostasis pathways is shown in Fig. 24.3 , and further details about individual hormones are described later.
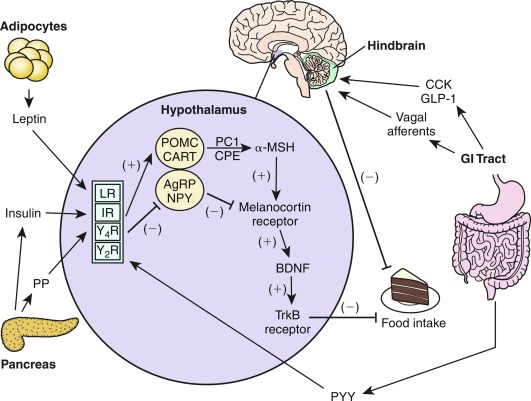
The Afferent System
Monitoring of metabolic status occurs in multiple regions of the body and assesses circulating forms of energy that are available for immediate use and fluctuate over the course of a meal cycle, as well as more constant stored forms of energy, namely fat. Direct detection of circulating forms of energy uses specific receptors for micronutrients or cellular detection systems, such as glycolytic flux generating varying amounts of adenosine triphosphate (ATP) depending on glucose availability. Indirect surveillance involves paracrine and endocrine hormones that reflect energy status and also stretch receptors in the GI tract that detect volume of material within the gut, nutritive or otherwise, rather than micronutrients specifically. The bloodstream and the autonomic nervous system, mainly the afferent vagus nerve, are the primary routes for transmission of metabolic information from the periphery to the CNS. The mediobasal hypothalamus (MBH, contains the ARC, ventromedial hypothalamic nucleus [VMH], and median eminence [ME]) and dorsal vagal complex (DVC, contains the nucleus of the solitary tract [NTS], the area postrema [AP], and dorsal motor nucleus of the vagus nerve [DMV]) of the brainstem medulla have a special fenestrated blood-brain barrier (BBB), allowing circulating hormones and micronutrients to diffuse into these brain regions for detection by specific receptors and sensing mechanisms. The DVC also receives sensory afferents from the vagal nerve, which innervates the GI tract from the esophagus to the colon and delivers information from chemical and mechanical sensors in the gut to the CNS.
Positive Regulation
In addition to intrinsic neurocircuitry that favors positive energy balance by default, currently, one additional positive regulatory signal from the periphery is known to enhance this drive.
Ghrelin. Ghrelin is an octanoylated 28-amino-acid peptide that is a ligand of the growth hormone secretagogue receptor (GHSR) and endogenously secreted by the stomach and duodenum in the fasting state. Ghrelin circulates within the bloodstream to the CNS, where it binds GHSRs in the hypothalamus and brainstem, accessed through fenestrations and selective transport across the BBB. Pituitary GHSR stimulation induces growth hormone (GH) release whereas VMH GHSR stimulation promotes positive energy balance through increased hunger, food intake, fat deposition, and reduced energy expenditure. To date, ghrelin is the only known peripherally produced orexigenic peptide hormone. Ghrelin secretion by the stomach terminates upon entry of nutrients into the stomach and intestines after meal consumption. Ghrelin rises in conjunction with increasing hunger, and ghrelin peaks at the conclusion of satiety when voluntary food consumption begins. Exogenous ghrelin infusion induces food intake, supporting the role of ghrelin in triggering meal initiation. Individuals with obesity compared with normal weight controls have a globally lower plasma ghrelin concentration but retain a similar circadian pattern of rise in ghrelin with fasting and decrease with meal intake, suggesting that ghrelin is responsive to, rather than a cause of, obesity. Weight loss because of dietary restriction leads to a rise in ghrelin whereas gastric bypass surgery results in decreased ghrelin, which may contribute to the differences in weight regain after weight loss by these different approaches. Importantly, ghrelin is activated from its prohormone form by binding of octanoic acid to a serine residue by ghrelin O-acetyltransferase (GOAT). The unacylated form is active as a homeostatic signal yet does not bind to the GH receptor.
Negative Regulation
Satiation, satiety, and expenditure of excess energy are achieved by negative regulatory elements that respond acutely to intake and to long-term energy stores.
Stretch Receptors. Ingested material entering the stomach applies pressure to the gastric wall although mechanosensitive receptors formed by vagal afferents whose cell bodies reside in the nodose ganglion and terminals lie within the NTS of the brainstem DVC. The stretch signal slows gastric emptying, which promotes retention of food in the stomach allowing satiation to be reached.
Glucose. Digestion of carbohydrates leads to liberation of glucose and other sugars, which are readily absorbed into the hepatic portal vein and taken up by liver hepatocytes. A significant portion of the incoming glucose also enters into systemic circulation, and blood glucose levels are detected by glucose-sensing neurons in the brain that are involved in regulation of glucose homeostasis, as well as energy balance. A subset of glucose-excited neurons responds to postprandial increases in glucose by stimulating energy expenditure through thermogenesis, whereas a subset of glucose-inhibited neurons respond to increased glucose by reducing the activity of hunger circuitry with the CNS.
Insulin and Amylin. Glucose entry into circulation from meal absorption triggers a proportional release of the peptide hormones insulin and amylin from pancreatic β-cells. Circulating insulin induces cellular uptake and utilization of glucose and glycogen synthesis, and enters the CNS through active transport across the BBB or through fenestrations in the BBB at the MBH and DVC to promote satiety and increasing energy expenditure. Amylin enhances satiation by slowing gastric emptying and mediates satiety through its actions in AP of the DVC.
Cholecystokinin. As food enters the proximal small intestine, enteroendocrine I-cells in the duodenum and proximal jejunum respond to nutrients, most strongly to long-chain free fatty acids (FFAs), by secreting CCK, an 8-amino-acid peptide with paracrine and endocrine hormone actions. Locally, CCK binds receptors in the pylorus to promote delayed gastric emptying, and stimulates vagal afferent receptors within the proximal intestine that transmit signals to the NTS and AP of the DVC to induce satiation. CCK also circulates within the bloodstream to the BBB-fenestrated MBH and DVC to suppress hunger and promote meal termination.
Peptide YY, Glucagon-Like Peptide-1, and Oxyntomodulin. As food enters the lower GI tract, enteroendocrine L-cells residing primarily in the ileum and colon become activated and secrete the peptide hormones peptide YY (PYY), GLP-1, and oxyntomodulin into circulation. The segment of intestine that secretes PYY, GLP-1, and oxyntomodulin is both downstream and considerably longer than that of the region where CCK is released, and thus the passage of food through this later segment of the gut leads to prolonged satiety between meals.
PYY circulates within the bloodstream to the brain as peptide fragments, predominantly as the 34-amino-acid PYY 3-36 , and binds type-2 neuropeptide Y receptors (Y2Rs), predominantly in the ARC, where it suppresses orexigenic NPY signaling and activates anorexigenic POMC signaling, together inducing satiation and satiety. PYY also acts in a paracrine fashion by locally activating intestinal afferent vagal neurons that transmit anorexigenic signals to the DVC.
GLP-1 is produced by prohormone convertase 1 (PC1)-mediated proteolytic cleavage of preproglucagon within L-cells, and acts on the stomach to slow gastric emptying, contributing to satiation through food retention. GLP-1 also circulates to the MBH and DVC where it activates its receptor to induce satiation and satiety. In addition to these effects on energy homeostasis, GLP-1 also acts as an incretin (enhancer of glucose-dependent insulin secretion) upon binding of GLP-1 receptors expressed in pancreatic β-cells, thereby enhancing insulin-mediated negative energy balance.
Oxyntomodulin is also produced through PC1-mediated proteolytic cleavage of preproglucagon as a separate product from GLP-1. Oxyntomodulin activates the GLP-1 receptor, but appears to act as a biased agonist that preferentially engages different intracellular signaling pathways from that of GLP-1, resulting in extended actions on satiation and satiety. Oxyntomodulin also activates the glucagon receptor, which stimulates energy expenditure through increased thermogenesis.
Pancreatic Polypeptide. Food ingestion stimulates, via CCK and vagal efferent signals delivered to the pancreas, the secretion of pancreatic polypeptide (PP) from pancreatic PP-cells into circulation. Circulating PP slows gastric emptying and mediates satiety by activating type-4 NPY receptors (Y4Rs) in the hypothalamus that increase anorexigenic POMC neuronal activity and decrease orexigenic NPY expression. Postprandial PP levels remain elevated for several hours, likely persisting into the postabsorptive phase and mediating satiety.
Bile Acids and Fibroblast Growth Factor 19. Bile acids are synthesized from cholesterol in the liver and are secreted into the duodenum after a meal, serving as lipid emulsifiers and signaling molecules of nutritional status via the G-protein–coupled receptor 19 (GPCR19) and farnesoid X-activated receptor (FXR). GPCR19 and FXR are expressed in the small intestine, liver, and adipose tissue. Activation of GPCR19 stimulates GLP-1 secretion and increases colonic peristalsis. Activation of FXR induces fibroblast growth factor 19 (FGF19) secretion into the bloodstream. FGF19 regulates lipid and glucose metabolism within the liver and also acts within the CNS to reduce food intake and increase energy expenditure.
Leptin. Adipocytes secrete the 167-amino-acid peptide hormone leptin in proportion to the amount of stored body fat. Typically, this secretion follows a circadian pattern with higher levels at night during sleep. However, when the postabsorptive phase progresses into extended fasting and subsequent starvation, leptin levels rapidly decrease, leading to potent food-seeking behavior. This striking effect indicates that an acute drop in leptin serves as a starvation signal and confirms leptin’s primary role as a protector of energy stores rather than as a preventer of obesity. Importantly, this acute effect is achieved regardless of basal leptin levels, thus starvation or extended fasting induces the same metabolic adaptations and behaviors in individuals with and without obesity. Leptin secretion is enhanced by markers of nutrient availability, including glucose, insulin, and cortisol, which all rise with meal intake, and is suppressed by catecholamines released with activation of the sympathetic nervous system (SNS, consistent with the “fight or flight” response requiring diversion of attention away from “rest and digest” behaviors). Leptin is a prerequisite signal of sufficient energy stores to permit initiation of high-energy processes, such as puberty and pregnancy. Programming of relative leptin concentrations by early caloric intake may be one mechanism that links early over nutrition with later obesity.
Leptin accesses the CNS through the BBB fenestrations of the MBH and DVC and is also actively transported across the BBB. Leptin’s primary site of action is the MBH but it also acts in other regions in the CNS and periphery. Leptin receptors are expressed by white adipocytes suggesting paracrine autoregulation, liver hepatocytes, pancreatic islet cells including insulin-secreting β-cells, neurons throughout the brain, and a portion of brain vascular endothelial cells (cerebrovascular cells) that form the BBB. The leptin receptor (a member of the cytokine receptor superfamily) has four isoforms, formed by differential messenger ribonucleic acid (mRNA) splicing: ObRa, an isoform with a shortened intracellular domain, which may function as a transporter; ObRb, the intact full-length receptor; ObRc, also with a short intracellular domain; and ObRe, without an intracellular domain, but which may function as a soluble receptor. Leptin receptor (LepR) activation leads to three primary neuronal signals. The first is the opening of an ATP–sensitive potassium channel, which hyperpolarizes the neuron and decreases its firing rate. The second is the activation of a cytoplasmic Janus kinase 2 (JAK2), which phosphorylates a tyrosine moiety on proteins of a family called signal transducers and activators of transcription (STAT-3). The phosphorylated STAT-3 translocates to the nucleus, where it promotes leptin-dependent gene transcription. However, leptin also activates the insulin receptor substrate 2/phosphatidyl inositol-3-kinase (IRS-2/PI3K) second messenger system, which increases neurotransmission of the central anorexigenic signaling pathway.
LepR activation in hypothalamic and brainstem regions suppresses the activity of orexigenic neurons and activates anorexigenic activity that increases energy expenditure. LepR activation increases SNS efferent tracts connected to adipocytes and acts to reduce leptin secretion, suggesting that leptin, like classic hormones, may be autoregulated by an endocrine feedback loop. Leptin also appears to have a role in regulating energy thermogenesis through reduction in the thermoregulatory tolerance of colder temperatures, in essence increasing the body’s thermostat to a higher temperature set-point. Thus when leptin levels are reduced during fasting, body temperature is not defended as strongly and thermogenic energy expenditure is therefore decreased to conserve energy stores.
Central Processing
The peripheral afferent signals outlined earlier reach the CNS and act primarily within the hypothalamus and brainstem, where they are integrated by a gated neural circuit, designed to promote net catabolic or anabolic effects (see Fig. 24.2 ).
Central Catabolic Signals
POMC/CART Neurons. The ARC houses neurons that coexpress POMC and CART. POMC is a peptide that is proteolytically cleaved by PC1 and carboxypeptidase E (CPE) to form different peptides depending on neuron type and location. Cleaved products include β-endorphin, adrenocorticotrophic hormone (ACTH), and, in ARC POMC-expressing neurons (ARC POMC ), α-melanocyte-stimulating hormone (α-MSH). Both overfeeding and peripheral leptin infusion induce the synthesis of POMC and α-MSH within the ARC. ARC POMC neurons are also directly activated by insulin, glucose, and serotonin. ARC POMC neurons are inhibited by ghrelin and AgRP neurons. α-MSH induces anorexia by binding to receptors within the paraventricular hypothalamic nucleus (PVN) and lateral hypothalamus (LHA). CART is a hypothalamic neuropeptide induced by leptin and reduced by fasting. Intrahypothalamic infusion of CART blocks appetite, whereas antagonism of endogenous CART increases caloric intake.
Melanocortin Receptors. The cleaved peptide products of POMC bind to and activate various 7-transmemberane G-protein–coupled melanocortin receptors (MCRs): MC1R in skin and hair stimulates production of the dark pigment, melanin; MC2R in the adrenal glands stimulates production of glucocorticoids; and melanocortin-4 receptor (MC4R) and melanocortin-3 receptor (MC3R) in the CNS induce negative energy balance. Activation of MC4R in the PVN and LHA results in a state of satiety, whereas intracerebroventricular (ICV) administration of MC4R antagonists in rodents stimulates feeding. MC4R-null mice display severe hyperphagia and obesity. MC3R-null mice display milder obesity and are not hyperphagic but appear to have higher feeding efficiency and greater fat partitioning.
Brain-Derived Neurotrophic Factor. Brain-derived neurotropic factor (BDNF) is an activity-dependent neurotrophin that regulates synaptic plasticity. BDNF has additionally been shown to play an important role in energy homeostasis as a downstream mediator of the leptin-melanocortin pathway. BDNF expression in the VMH is regulated by nutritional state and MC4R signaling. Selective deletion of BDNF from VMH and dorsomedial hypothalamic nucleus (DMH) of adult mice causes hyperphagia and obesity, whereas infusion of BDNF into VMH of wild-type (WT) rats reduces food intake and increases energy expenditure. In the anterior PVN, BDNF suppresses food intake and increases locomotor activity, whereas in medial and posterior PVN, BDNF stimulates thermogenesis through increased SNS outflow. BDNF haploinsufficiency in both humans and rodents is associated with obesity, which in mice, can be prevented with pair-feeding, suggesting hyperphagia as the primary driver of weight gain, BDNF dysfunction may also contribute to overeating behaviors found in Prader-Willi syndrome (PWS) and Smith-Magenis syndrome (SMS), as well as in common eating disorders. The BDNF Val66Met polymorphism, which impairs activity-dependent BDNF secretion, is linked to binge episodes in bulimia nervosa and binge eating disorder, and BDNF hypermethylation is associated with bulimia nervosa. The intronic BDNF rs12291063 variant (homozygous in ~ 10% of individuals with African ancestry) is associated with reduced VMH BDNF expression and increased adiposity. Together these observations indicate that BDNF insufficiency may underlie common, as well as rare causes of overeating behaviors.
Tropomyosin-Related Kinase B Receptor. Tropomyosin-related kinase B (TrkB) is encoded by the NTRK2 gene and is the cognate receptor for BDNF. Chemogenetic activation of TrkB-expression neurons in the DMH suppresses feeding during the dark cycle when mice are physiologically hungry, whereas chemogenetic inhibition of these neurons promoted feeding during the light cycle when mice are physiologically satiated. Selective Ntrk2 deletion in the DMH of adult mice induces hyperphagia, decreased energy expenditure, and obesity.
Norepinephrine. Norepinephrine ( NE) neurons in the locus coeruleus synapse on VMH neurons to regulate food intake. In rodents, the actions of NE on food intake seem paradoxical, as intrahypothalamic NE infusion stimulates food intake through effects on central α 2 – and β-adrenergic receptors, whereas central infusion of α 1 -agonists markedly reduces food intake.
In human studies using molecular neuroimaging, NE transporter availability was negatively associated with perceptions of hunger and the strength of this association was stronger among individuals with obesity compared with normal-weight controls, suggesting that NE has a role in modulating hunger.
Serotonin (5-hydroxytryptamine). 5-Hydroxytryptamine (5-HT) has been implicated in the perception of satiety based on many lines of evidence: (1) injection of 5-HT into the hypothalamus increases satiety, particularly with respect to carbohydrate ; (2) central administration of 5-HT 2c receptor agonists increase satiety, whereas antagonists induce feeding ; (3) administration of selective 5-HT reuptake inhibitors induce early satiety ; (4) leptin increases 5-HT turnover ; and (5) the 5-HT 2c R-KO mouse exhibits increased food intake and body weight. The role of 5-HT in the transduction of the satiety signal may have both central and peripheral components, as intestinal 5-HT is secreted into the bloodstream during a meal, where it may have an impact on GI neuronal function and muscle tone, and may bind to 5-HT receptors in the NTS (discussed previously) to promote satiety. Molecular neuroimaging in humans suggests that obesity is driven by decreased serotonin-mediated homeostatic feedback in response to food intake.
Central Anabolic Signals
AgRP/NPY Neurons. NPY and AgRP colocalize to a different set of neurons within the ARC, immediately adjacent to those expressing POMC/CART. The ARC houses the only population of neurons that express AgRP, and the majority of these neurons coexpress NPY. These two orexigenic peptides are secreted from nerve terminals as peptide neurotransmitters, in addition to the inhibitory small-molecule neurotransmitter gamma-amino butyric acid (GABA). AgRP is the human homolog of the protein agouti, which is present in abundance in the Agouti yellow mouse (A y -a). AgRP is an endogenous competitive antagonist of all MCRs, accounting for the yellow color in these mice because of its inhibitory actions at the MCR1 receptors of the fur. AgRP also antagonizes MC4R and MC3R, thereby attenuating the ability of α-MSH to suppress appetite and fat deposition. AgRP neurons also suppress SNS-mediated “browning” of white adipose tissue which would otherwise increase energy expenditure, thus reducing thermogenesis and promoting positive energy balance. AgRP neurons are directly activated by the orexigenic hormone ghrelin ; dopamine, which may originate from dopamine neurons residing within the ARC ; and several glutamatergic projections from other hypothalamic nuclei including the DMH (which exhibits leptin BBB transport and activity) and the PVH. AgRP neurons are inhibited by leptin, insulin, PYY, and by GABAergic projections from the DMH and other arcuate neurons.
NPY has numerous functions within the hypothalamus, including initiation of feeding, regulation of gonadotropin secretion, and modulation of adrenal responsiveness. NPY acts as an orexigen and it also stimulates adipogenesis. ICV infusion of NPY in rats rapidly leads to hyperphagia, energy storage, and obesity, mediated through Y 1 and Y 5 receptors. Fasting and weight loss increase NPY expression in the ARC, accounting for increased hunger, whereas PYY 3-36 (through Y 2 receptors) and leptin decrease NPY mRNA.
Melanin-Concentrating Hormone. Melanin-concentrating hormone (MCH) is a 17-amino-acid orexigenic peptide expressed in the zona incerta and LHA. MCH-knockout mice are hypophagic and lean, whereas transgenic MCH-overexpressing mice develop obesity and insulin resistance. ICV administration of MCH stimulates food intake, similar to that seen with NPY administration. Interestingly, endogenous MCH circulation within the cerebrospinal fluid (CSF) may in fact represent an alternative neural communication mechanism in the regulation of food intake with rise in CSF concentrations of MCH driving the initiation of food intake.
Orexins A and B. Orexin A and B are orexigenic peptides (33 and 28 amino acids in length, respectively) produced within the hypothalamus and modulate both energy balance and autonomic function in mice. Orexins stimulate release of NPY, corticotropin-releasing hormone (CRH), and SNS outflow leading to increased food intake, wakefulness, blood pressure, and energy expenditure. The orexins also appear to bridge the homeostatic and non-homeostatic mechanisms that regulate food intake and may play a role in reward-based learning and memory. Orexin neurons in the LHA process the hedonic aspects of food and drugs of abuse, whereas orexin neurons in the perifornical and DMH regulate arousal and stress response.
Endocannabinoids. Tetrahydrocannabinol, the main psychotropic component of marijuana, has long been known to stimulate food intake. The endogenous endocannabinoid (EC) receptor, CB 1 , is expressed in corticotropin-releasing hormone (CRH) neurons in the PVN, in CART neurons in the VMN, and in MCH- and orexin-positive neurons in the LHA and perifornical region. Fasting and feeding are associated with high and low levels of ECs in the hypothalamus, respectively. For example, CB 1 receptor-knockout mice have increased CRH and reduced CART expression. In the leptin-deficient ob/ob mice, hypothalamic EC levels are increased, whereas leptin infused intravenously reduces these levels, indicating that a direct negative control is exerted by leptin on the EC system. Glucocorticoids increase food intake by stimulating EC synthesis and secretion, whereas leptin blocks this effect. Also the presence of CB1 receptors on afferent vagal neurons suggests that EC may be involved in mediating satiety signals originating in the gut.
The Efferent System
The MCRs in the PVN and LHA transduce the anorexigenic and orexigenic information coming from the VMH, to modulate activity of the SNS, and the efferent vagus, which promotes energy storage ( Fig. 24.4 ). In this way, peripheral energy balance can be modulated acutely to provide requisite energy for metabolic needs, and store the rest.
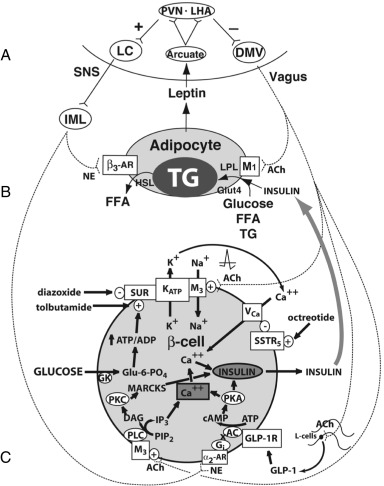
The Sympathetic Nervous System and Energy Expenditure
Anorexigenic pressure increases energy expenditure through activation of the SNS. For instance, leptin administration to leptin-deficient ob/ob mice promotes increased brown adipose tissue lipolysis, thermogenesis, renovascular activity, and increased movement, all associated with increased energy expenditure and enhanced weight loss. Similarly, insulin administration acutely increases SNS activity in normal rats and in humans. The SNS increases energy expenditure in four ways: (1) by innervating the hypothalamus and appetite centers in the medulla to reduce appetite, (2) by increasing thyroid-stimulating hormone (TSH) secretion to increase thyroid hormone release and energy expenditure, (3) by innervating skeletal muscles to increase energy expenditure, and (4) by innervating β 3 -adrenergic receptors in white adipose tissue to promote lipolysis.
Activation of the SNS increases energy expenditure by the skeletal muscle, by activating β 2 -adrenergic receptors, which in turn increase the expression of numerous genes in skeletal muscle, especially those involved in carbohydrate metabolism. SNS activation stimulates glycogenolysis, incites myocardial energy expenditure, increases in glucose and fatty acid oxidation, and increases protein synthesis.
Activation of the SNS in rodents stimulates the β 3 -adrenergic receptor of brown adipose tissue to promote lipolysis. In humans, activation of the β 3 -adrenergic receptor increases cyclic adenosine monophosphate (cAMP), which activates protein kinase A (PKA). PKA acts in two separate molecular pathways to increase energy expenditure. First, PKA phosphorylates the cAMP response element binding protein (CREB), which induces expression of peroxisome proliferator-activated receptor (PPAR)γ-coactivator-1α (PGC-1α). PGC-1α then binds to enhancer elements on the uncoupling protein-1 ( UCP1 ) gene, which increases the expression and activity of uncoupling proteins (UCPs) 1 and 2. UCPs reduce the proton gradient across the inner membranes of mitochondria, thereby diverting protons from storage in the form of ATP to heat production. Originally, UCPs were discovered in brown adipose tissue and were found to be responsible for thermogenesis. UCP1 is an inner membrane mitochondrial protein that uncouples proton entry from ATP synthesis; therefore UCP1 expression dissipates energy as heat, thus reducing the energy efficiency of the adipose tissue. However, UCP2 has been found in most tissues and UCP3 in skeletal muscle. Second, PKA activation activates the enzyme hormone-sensitive lipase (HSL), which is responsible for lipolysis of intracellular triglyceride to its component FFAs. The FFAs also induce UCP1, further increasing energy expenditure. The FFAs released from the adipocyte also travel to the liver where they are used for energy by being metabolized into two-carbon fragments. Lipolysis reduces leptin expression; thus a negative feedback loop is achieved between leptin and the SNS (see Fig. 24.4 ).
The Efferent Vagus and Energy Storage
In response to declining levels of leptin or persistent orexigenic pressure, the LHA and PVN send efferent projections residing in the medial longitudinal fasciculus to the DMV, activating the efferent vagus. The efferent vagus opposes the SNS by promoting energy storage in four ways: (1) by slowing the heart rate, myocardial oxygen consumption is reduced; (2) the vagus nerve promotes alimentary peristalsis, pyloric opening, and energy substrate absorption; (3) through direct effects on the adipocyte, the vagus nerve promotes insulin sensitivity to increase the clearance of energy substrate into adipose tissue; and (4) through effects on the β-cells, the vagus increases postprandial insulin secretion, which promotes energy deposition into adipose tissue.
Retrograde tracing of white adipose tissue reveals an abundance of efferents originating at the DMV. These efferents synapse on the M 1 muscarinic receptor on the adipocyte, which increases insulin sensitivity of the adipocyte. Denervation of white adipose tissue results in a reduction of glucose and FFA uptake, and an induction of HSL, which promotes lipolysis—both of which reduce the efficiency of insulin-induced energy storage. Thus vagal modulation of the adipocyte augments storage of both glucose and FFAs by improving adipose insulin sensitivity (see Fig. 24.4 ).
The DMV also sends efferent projections to the β-cells of the pancreas. This pathway is responsible for the “cephalic” or preabsorptive phase of insulin secretion, which is glucose independent and can be blocked by atropine. Overactive vagal neurotransmission increases insulin secretion from β-cells in response to an oral glucose load through three distinct but overlapping mechanisms (see Fig. 24.4 ):
- 1.
Vagal firing increases acetylcholine availability and binding to the M 3 muscarinic receptor on the β-cell, which is coupled to a sodium channel within the pancreatic β-cell membrane. As glucose enters the β-cell after ingestion of a meal, the enzyme glucokinase phosphorylates glucose to form glucose-6-phosphate, increasing intracellular ATP, which induces closure of the ATP-dependent potassium channel. Upon channel closure, the β-cell experiences an ATP concentration-dependent β-cell depolarization and the opening of a separate voltage-gated calcium channel within the membrane. Intracellular calcium influx increases acutely, which results in rapid insulin vesicular exocytosis. Concomitant opening of the sodium channel by vagally mediated acetylcholine augments β-cell depolarization, which in turn augments the intracellular calcium influx and results in insulin hypersecretion.
- 2.
Vagally mediated acetylcholine increases phospholipases A 2 , C, and D within the β-cell, which hydrolyze intracellular phosphatidylinositol to diacylglycerol (DAG) and inositol triphosphate (IP 3 ). DAG is a potent stimulator of protein kinase C (PKC), which phosphorylates myristoylated alanine-rich protein kinase C substrate (MARCKS), which then binds actin and calcium-calmodulin and induces insulin vesicular exocytosis. IP 3 potentiates the release of calcium within β-cells from intracellular stores, which also promotes insulin secretion.
- 3.
The vagus also stimulates the release of GLP-1 from intestinal L-cells, which circulates and binds to a GLP-1 receptor within the β-cell membrane. Activation of this receptor induces a calcium-calmodulin-sensitive adenyl cyclase, with conversion of intracellular ATP to cAMP, which then activates PKA. PKA causes both the release of intracellular calcium stores and the phosphorylation of vesicular proteins, each contributing to an increase in insulin exocytosis.
In the efferent pathway, insulin is responsible for shunting blood-borne nutrients into adipose for storage. Indeed, the primary hormonal signal for adipogenesis is insulin. Within the adipocyte, insulin increases: (1) glucose transporter 4 (GLUT 4) expression, (2) acetyl-CoA carboxylase, (3) fatty acid synthase, and (4) lipoprotein lipase. Thus the net effect of insulin on the adipocyte is the rapid clearance and storage of circulating glucose and lipid thereby promoting energy storage.
Non-homeostatic regulation of energy balance
The non-homeostatic determinants of energy balance are higher order functions that integrate cognitive functioning and environmental cues that prompt nonnutritive reasons for eating. Homeostatic feeding is needed for survival, whereas hedonic feeding is driven by the reward aspects of palatable food. In contrast to rodents or other mammals, hedonistic drives in humans may override homeostatic drives and dictate eating behavior.
The Nucleus Accumbens and the Hedonic Pathway of Food Reward
The hedonic pathway comprises the ventral tegmental area (VTA) and the nucleus accumbens (NA), with inputs from various components of the limbic system, including the striatum, amygdala, hypothalamus, and hippocampus. These pathways also mediate the hedonic response to drugs of abuse, such as nicotine and morphine. In fact, administration of morphine to the NA increases food intake in a dose-dependent fashion. When functional, the hedonic pathway helps curtail food intake in situations where energy stores are replete; however, when dysfunctional, this pathway can increase food intake leading to obesity.
The VTA appears to mediate feeding on the basis of palatability rather than energy need. The dopaminergic projection from the VTA to the NA mediates the motivating, rewarding, and reinforcing properties of various stimuli, such as food and addictive drugs. Leptin and insulin receptors are expressed in the VTA, and both hormones have been implicated in modulating rewarding responses to food and other pleasurable stimuli. For instance, fasting and food restriction (when insulin and leptin levels are low) increase the addictive properties of drugs of abuse, whereas ICV leptin can reverse these effects. In rodent models of addiction, increased addictive behavior (and pleasurable response from a food reward), as measured by dopamine release and dopamine receptor signaling, is greater after food deprivation. In humans with leptin deficiency, alterations in activity in the NA can be seen using functional magnetic resonance imaging (MRI) scanning, and these changes subside with administration of exogenous leptin. Acutely, insulin increases expression and activity of the dopamine transporter, which clears and removes dopamine from the synapse; thus acute insulin exposure blunts the reward of food. Furthermore, insulin appears to inhibit the ability of VTA agonists (e.g., opioids) to increase intake of sucrose. Finally, insulin blocks the ability of rats to form a conditioned place preference association to a palatable food. However, insulin resistance of this pathway may lead to increased reward perception of food by way of reduced dopamine clearance from the synapse and prolongation of the postsynaptic hedonistic response.
One question that has garnered increasing interest is whether any macronutrient has addictive properties. In animal studies, sugar has been shown to induce the four criteria for addiction: (1) bingeing, (2) withdrawal, (3) craving, and (4) cross-sensitization with other drugs of abuse. Within fast food, sugar and caffeine satisfy the criteria presented in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) for dependence in humans. However, the question of whether food addiction exists, and whether it can explain patients with obesity, remains contested, although a recent systematic review of the literature supports this notion of “highly-palatable food addiction” based on the criteria of impaired control, social impairment, risky use, and tolerance/withdrawal, at least as a paradigm for consideration in treatment strategies.
The Amygdala and the Stress Response
The VMH and VTA-NA mediate satiety when energy stores are replete, but they appear to be easily overridden by amygdala activation and resultant stress, a state of physiologic insulin resistance ( Fig. 24.5 ). Numerous lines of evidence suggest that the stress glucocorticoids corticosterone (in the rodent) or cortisol (in the human) are essential for the full expression of obesity, which helps to explain the disruptive role of stress in weight regulation.
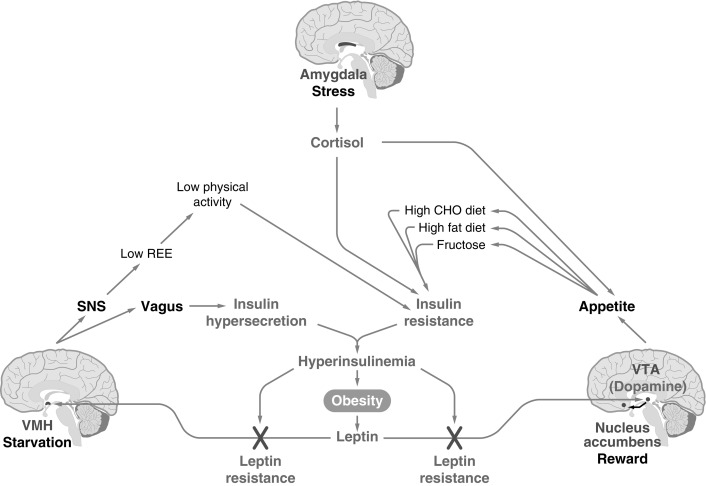
Stress and glucocorticoids are integral in promoting adiposity and the metabolic syndrome. Adrenalectomized rats maintained pharmacologically with high levels of corticosterone demonstrate that exogenous fat intake is directly proportional to circulating corticosterone concentrations, whereas amygdala activation by stress is dampened by the ingestion of energy-dense food. In intact rats, corticosterone stimulates eating, particularly of high-fat food, and in humans, cortisol administration increases food intake. Human research shows increased caloric intake of “comfort foods” (i.e., those with high energy density) after acute stress, and that the stress response contributes to leptin resistance (discussed later). Several studies in children have observed relationships between stress and unhealthy dietary practices, including increased snacking, and an elevated risk for problems with weight during adolescence and adulthood. In a controlled study of 9-year-old children who scored high on dietary restraint and who felt more stressed by laboratory challenges tended to eat more comfort food. Adverse childhood experiences are also associated with later development of obesity and cardiometabolic risk factors suggesting a role of stress in longitudinal weight gain and metabolic health.
Dysregulation of energy balance
Leptin Resistance
Most children with obesity have high leptin levels but do not have receptor mutations, manifesting what is commonly referred to as functional leptin resistance . Leptin resistance prevents exogenous leptin administration from promoting weight loss. The response to most weight-loss regimens plateaus rapidly because of the rapid fall of peripheral leptin levels which induces a “starvation response” immediately, regardless of baseline values, and potentially because of reaching a personal “leptin threshold,” which is likely genetically determined. Leptin decline causes the VMH to sense a reduction in peripheral energy stores, which modulates a decrease in resting energy expenditure (REE) to conserve energy, analogous to a starvation response, but occurring at elevated leptin levels.
The cause of leptin resistance is unknown, but it may have several etiologies. Leptin crosses the BBB via a saturable transporter, which limits the amount of leptin reaching its receptor in the VMH ; this transporter operates more efficiently at lower levels of leptin, while preventing increased signaling at higher levels. Activation of the leptin receptor induces the intraneuronal expression of suppressor of cytokine signaling-3 (SOCS-3), which limits leptin signal transduction in an autoregulatory fashion. Because the presence of hyperleptinemia has been shown to be a prerequisite for development of leptin resistance, it has been postulated that leptin-induced expression of leptin signaling inhibitors may be an initial step in the process. Other studies suggest that obesity itself induces hypothalamic inflammation, gliosis, and endoplasmic reticulum (ER) stress that impair responsiveness to leptin.
The standard method for producing insulin resistance and obesity in rodents is a high-fat diet. Dietary fat promotes leptin resistance through its effects on hypertriglyceridemia, which limits access of peripheral leptin to the VMH, and also by interfering with leptin signal transduction upstream of STAT-3, its primary second messenger. One likely modulator of this pathway is the enzyme PI3K, which is the downstream effector of insulin action in POMC neurons and which appears to account for the effects of dietary fat on leptin resistance and obesity.
Two clinical paradigms have been shown to improve leptin sensitivity. After weight loss through caloric restriction, exogenous administration of leptin can then increase REE back to baseline and permit further weight loss, suggesting that the weight loss itself improves leptin sensitivity. Second, suppression of insulin correlates with improvement in leptin sensitivity and promotes weight loss, suggesting that hyperinsulinemia promotes leptin resistance by interfering with leptin signal transduction in the VMH and VTA. Indeed, insulin reduction strategies can effectively promote weight loss in children with hyperinsulinemia by improving leptin sensitivity.
Counterregulatory Mechanisms That Oppose Weight Loss
Because the homeostatic system for energy balance was developed to protect against starvation, body fat stores are avidly protected. Therefore dietary restriction, even before the onset of weight loss, triggers counterregulatory mechanisms to oppose the perceived threat of starvation, regardless of baseline adipose tissue stores. Gastric secretion of ghrelin acutely rises, which increases pituitary GH release, to stimulate lipolysis to provide energy substrate for catabolism. Ghrelin stimulates NPY/AgRP to antagonize α-MSH/CART. Decline of leptin reduces α-MSH/CART as well. This leads to decreased MC4R and MC3R occupancy leading to reduced anorexigenic and catabolic signaling with a net effect of increased feeding behavior and higher energy efficiency (with reduced fat oxidation). Appetite proportionally increases leading to food consumption above baseline by approximately 100 kCal/d per kilogram of lost weight. Meanwhile, total and resting energy expenditures decline in an attempt to conserve energy. Specifically, UCP1 levels within adipose tissue decline as a result of decreased SNS activity. In spite of decreased SNS tone at the adipocyte, there is clearly an obligate lipolysis (because of insulin suppression and upregulation of HSL), which is necessary to maintain energy delivery to the musculature and brain in the form of liver-derived ketone bodies. In addition, in the weight-reduced state, vagal tone is increased to slow the heart rate and myocardial oxygen consumption, increase β-cell insulin secretion in response to glucose, and increase adipose insulin sensitivity—all directed to increase energy storage. These counterregulatory mechanisms together serve to drive weight regain and can even persist for years after the initial onset of weight loss, therefore rendering maintenance of reduced body weight exceedingly challenging especially in light of the feed-forward mechanisms described later. In other words, the metabolic adaptations aimed at conserving energy and returning to the body weight before weight loss are maintained for years following the weight loss and achievement of weight plateau rendering the individual prone to weight gain. Upon comparison of two individuals with similar weight and body composition, a weight-stable patient and a patient who lost weight to achieve this measurement, to maintain the current body weight—the patient who lost weight will have to consume less energy and spend more energy in comparison with the weight-stable counterpart.
Feed-Forward Mechanism That Promote Weight Gain
Feed-forward signaling bridges homeostatic and hedonic mechanisms of appetite regulation, a concept developed based on the observation that a hungry mouse will have appropriately elevated AgRP neuronal firing in the fasted state but these AgRP neurons are then acutely suppressed when the mouse is presented with food, even before the onset of eating. In addition, gut signals are also released that anticipate the imminent arrival of ingested food, setting in motion, digestive processes before actual food intake. Because hunger is an aversive experience, rapid reduction in AgRP firing and the sudden removal of hunger sensations induce an acute reward experience, perhaps serving as a positive reinforcer of the environmental cue of food availability. For example, readily visible and appealing packaging of processed foods encourages hedonistic consumption and establishes a learned behavior that prompts subsequent return to the environment where such foods were available. Along the same lines, fast food restaurants and advertising targeting youth rely heavily on connecting images of food with other pleasurable stimuli (toys, games, fun, etc.), further compounding the reward effect of already highly palatable food. This feed-forward mechanism has potential implications as we consider the role of the built environment and its role in promoting food consumption.
Energy excess—obesity
The rise in the prevalence of obesity in children and adolescents is one of the most alarming public health issues facing the world today. Although the rise in the prevalence of obesity in children and adolescents seems to have leveled in some parts of the world, many others, especially developing countries and migrant populations, are still experiencing a steady increase. Obesity is associated with significant health problems in children and is an early risk factor for much of adult noncommunicable disease morbidity and mortality, and an important factor in increasing healthcare expenditures. Childhood obesity tends to track into adulthood, and those who continue to have obesity as adults have a significant risk for the development of obesity-driven morbidity with an excess risk associated with the length of exposure to obesity. In contrast, children with obesity who lost weight and became nonobese adults do not have an increased risk for such morbidity. These observations identify obesity in early childhood as a major window of opportunity for obesity prevention efforts with a potential lifelong impact.
Definition
The theoretical definition of obesity is a degree of somatic overweight that affords detrimental health consequences. Based on morbidity and mortality statistics, and with a desire to prevent future risk of morbidity, we practically define obesity as a statistical magnitude of overweight for a population, keeping in mind that morbidity and mortality vary with degree of overweight in different racial, ethnic, and socioeconomic groups.
The majority of obesity in adulthood has its origins in childhood, making obesity a pediatric concern and the prevention and treatment of obesity a pediatric goal. Body mass index (BMI) is also the accepted marker in children. In childhood, comparison of BMI with normal curves for age allows for categorization of BMI above the 85 th percentile as overweight, and above the 95 th percentile as obesity ( Fig. 24.6 A and B ). The World Health Organization (WHO) categorizes adult overweight into four subgroups based on BMI (weight [kg] ÷ height [m] 2 ): BMI 25 to 30 (overweight); BMI 30 to 5, Grade 1 (moderate obesity); BMI 35 to 40, Grade 2 (severe obesity); and BMI over 40, Grade 3 (morbid obesity). A similar degree of obesity categorization can also be used in the pediatric population using BMI centiles where the 95 th centile for age and sex is used as a reference point and 100% to 120% of the 95 th percentile for age and sex is grade 1, 120% to 140% is grade 2, and over 140% is grade 3. Such categorization demonstrates that cardiometabolic risk increases with rising degrees of obesity.
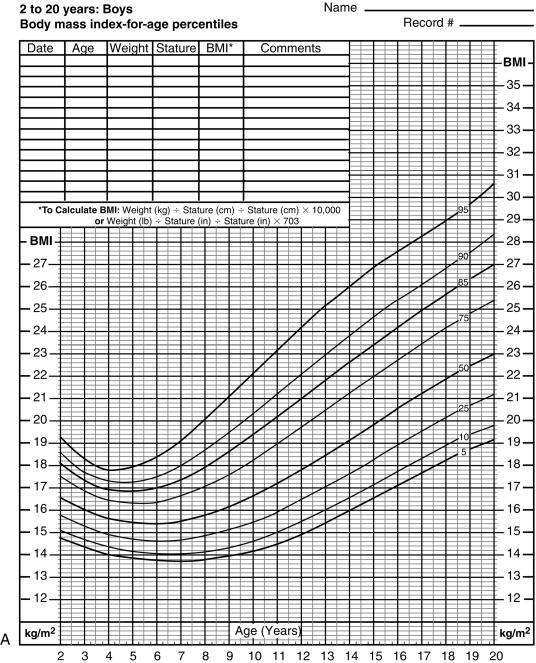
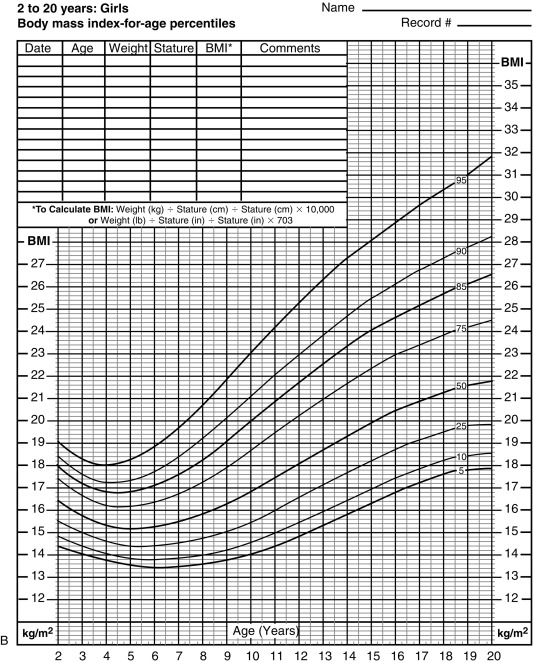
Although BMI is the standard indicator of obesity for statistical purposes and within populations, it should be noted that BMI does not take into account body composition parameters such as total body fat, (as well as both subcutaneous and visceral fat), muscle, and bone. Furthermore, BMI in children is age, sex, and puberty dependent, thus BMI z-score is a more accurate assessment of childhood adiposity. Lastly, waist circumference (an indirect measure of intraabdominal visceral fat) has emerged as a more accurate indicator of metabolic disturbance in children. These limitations of BMI indicate that it is a useful index for population and epidemiologic studies yet should be used with caution when assessing an individual child in the clinical setting.
Prevalence and Epidemiology
The prevalence of childhood obesity in the United States has increased dramatically during the past 30 years, and continues to do so, although the comparison of longitudinal and cross-sectional data is difficult because of different definitions and measurement parameters between epidemiologic studies. The most recent estimates of obesity prevalence and trends in the United States are based on data from the 2011 to 2014 National Health and Nutrition Examination Survey (NHANES V). NHANES demonstrates that the epidemic of childhood obesity in the United States seems to have stabilized in some age groups but not in others. Overall, in 2013 to 2014, 9.4% (95% confidence interval [CI], 6.8–12.6) of infants and toddlers and 17% (95% CI, 15.5–18.6) of children and adolescents from 2 through 19 years of age had obesity. Of note, the prevalence of extreme obesity (> 120% of 95 th percentile for age and sex) was 5.8% (95% CI, 4.9– 6.8). Trend analyses over a 25-year period indicated a significant increase in obesity prevalence among children aged 2 to 5 years between 1988 and 1994 and 2003 and 2004 that slightly declined in 2013 to 2014 (7.2%, 13.9%, and 9.4%, respectively). Among children aged 6 to 11 years old, the prevalence of obesity increased from 1988 to 1994 to 2007 to 2008 and remained stable in 2013 to 2014 (11.3%, 19.6%, and 17.4%, respectively). Among adolescents 12 to 19 years of age, the prevalence of obesity significantly increased from 1988 to 1994 to 2013 to 2014 (10.5% and 20.6%, respectively, P < .001). Of note, the prevalence of extreme obesity increased among children 6 to 11 years old between 1988 and 1994 to 2013 and 2014 (3.6% and 4.3%, respectively, P = .02) and among adolescents aged 12 to 19 years (2.6% and 9.1%, respectively, P < .001). Importantly, no significant trends were observed between 2005 and 2006 and 2013 and 2014. The practical implication of these trends is for example in 1988 to 1994, the 95 th percentile of BMI among 17-year-old males was 31.5 kg/m 2 (i.e., 5% of males had a BMI > 31.5), and in 2011 to 2014 the 95 th percentile was 36.2 kg/m 2 (i.e., 5% of males had a BMI > 36.2). Thus between 1988 and 1994 and 2013 and 2014, the prevalence of obesity increased until 2003 to 2004 and then decreased in children aged 2 to 5 years, increased until 2007 to 2008 and then leveled off in children aged 6 to 11 years, and increased among adolescents aged 12 to 19 years. In 2013 to 2014, 17.4% of children met criteria for class I obesity, including 6.3% for class II and 2.4% for class III. A clear, statistically significant increase in all classes of obesity continued from 1999 through 2014. In the United States, obesity and severe obesity among children significantly increased with greater age and lower education of household head, and severe obesity increased with lower level of urbanization. Compared with non-Hispanic white youth, obesity and severe obesity prevalence were significantly higher among non-Hispanic black and Hispanic youth. Severe obesity, but not obesity, was significantly lower among non-Hispanic Asian youth than among non-Hispanic white youth. Lastly, projections argue that by 2030, 42% of American adults will have obesity.
Global Prevalence
Obesity has overtaken acquired immunodeficiency syndrome and malnutrition as the number one public health problem in the world. The global prevalence of childhood obesity has been increasing worldwide at an alarming rate during the past 20 years. Rates have increased 2.7 to 3.8-fold over 29 years in the United States, 2.0 to 2.8-fold over 10 years in England, 3.4 to 4.6-fold over 10 years in Australia, and 3.4 to 3.6-fold over 23 years in Brazil. European data, using slightly different obesity cutoff definitions (a childhood BMI corresponding to > 25 and 30 kg/m 2 in adults signifying overweight and obesity, respectively), suggests that among European countries, prevalence of overweight/obesity combined ranges between 16% and 22% whereas that of obesity ranges between 4% and 6% (corresponding to 2.9–4.4 million children with obesity in the European continent). Rapid increases in the prevalence of overweight schoolchildren are being seen in all European countries for which data are available. The numbers indicate a lag of 10 to 15 years behind the United States. Using data from the mid-70s to 2016 in 200 countries, it was shown that trends in mean BMI have recently flattened in northwestern Europe and the high-income English-speaking and Asia-Pacific regions for both sexes, southwestern Europe for boys, and central and Andean Latin America for girls. In contrast, the rise in BMI has accelerated in east and south Asia for both sexes, and southeast Asia for boys. In developed countries, the urban poor are more susceptible for developing obesity, presumably because of poor dietary practices and limited opportunity for physical activity. In contrast, obesity is more frequent in upper socioeconomic class of developing countries, probably because of a nutrition transition to a more Western diet with more energy-dense items consisting of higher fats and sugar, which tend to be more palatable at a lower cost. This may be also caused by specific properties of processed food, which may promote leptin resistance.
Racial and Ethnic Considerations
The NHANES surveys only list prevalence among non-Hispanic whites, non-Hispanic blacks, and Asians, despite the fact that Native Americans, Pacific Islanders, and other racial/ethnic groups are experiencing rapid increases in obesity prevalence as well. Across racial groups, there is a marked dichotomy in the prevalence, and in the rate of increase of childhood obesity. For instance, the prevalence among African American (24.4%), Hispanic (21.7%) and Mexican American adolescents (22.2%) is significantly higher than among white adolescents (15.6%). Importantly, the prevalence of severe obesity (BMI > 97 th percentile) among African American (18.5%), Hispanic (15.2%) and Mexican American adolescents (15.2%) by far exceeds that of non-Hispanic white adolescents (10.5%). The rate of increase in the prevalence of obesity among African American and Hispanic adolescents almost doubled between 1988 and 1994 and 1999 and 2000, from 13.4% to 23.6% in African Americans, and from 13.8% to 23.4% in Hispanics. The 1994 Pediatric Nutrition Surveillance System (PedNSS) indicated that 12% of 2- to 4-year-old Native American children were overweight, which is similar to Hispanic children at the same age (12%) but much higher than white children (6%). The prevalence of overweight at 5 to 6 years in Native Americans is twice that in US youth in general, and the prevalence of obesity is even 3 times higher. Overall, both American Indian and Alaska Native children and adolescents have a greater prevalence of obesity compared with US children overall. Among infants and toddlers less than 2 years of age, the prevalence of obesity is highest in African Americans (18.5%), as compared with 10.1% in non-Hispanic whites and 13.7% in Hispanics. It is possible that different dietary practices may account for some of these differences. For instance, a study of 2-year-old Latino children in California correlated obesity with early consumption of sugar-sweetened beverages.
Within racial populations, ethnic variability in the prevalence of childhood obesity has also been noted. Only 25% of first-generation Hispanic adolescents were overweight based on BMI in the 85 th percentile or higher, as compared with 32% of second- and third-generation Hispanics. The prevalence of overweight in Asian American adolescents in this study was 20.6%, with comparable prevalence among Filipinos (18.5%) and Chinese (15.3%). Again, only 12% of first-generation Asian Americans were overweight, compared with 27% and 28%, of second and third generations, respectively. In Native Americans, there is great variation in the prevalence of obesity from 12% to 77%, based on tribes, age groups, measurement tools, and cut off values, among the studies performed between 1990 and 2000. These studies indicate that obesity in Native Americans begins very early in childhood.
Predictive Factors
The higher the BMI during childhood, the more likely adult obesity will manifest. In general, children with a BMI in the 95 th percentile or higher have a very high risk for adult obesity. Obesity in adolescence is a primary risk factor for obesity in adulthood, with an increased odds ratio from 1.3 for obesity at 1 to 2 years of age to 17.5 for obesity at 15 to 17 years of age. The strongest predictor of adolescent obesity is rapid weight gain between 2 and 6 years of age. The change of BMI during and after adolescence is the most important predictive variable for adult obesity. Children and adolescents with BMI in the 95 th percentile or higher have a 62% to 98% chance of having obesity at 35 years of age, with a 50% chance in males aged 13 years or older and 66% chance in girls age 13 years or older. Importantly, an elevated BMI in adolescence (even one that is considered well within the “normal” range) constitutes a substantial risk factor for obesity-related disorders in midlife. Although the risk of diabetes is mainly associated with increased BMI close to the time of diagnosis, the risk of coronary heart disease is associated with an elevated BMI both in adolescence and in adulthood.
The age of adiposity rebound, that is, the point of the BMI nadir before body fatness begins to rise (between 5 and 6 years of age), typically more pronounced in girls (see Fig. 24.6 A and B), is also an important predictor for adult obesity. Children with early adiposity rebound have a fivefold greater chance of having obesity as adults, compared with those with late adiposity rebound. At the age of adiposity rebound, children already overweight have a sixfold greater risk for adult obesity, as compared with lean children. Weight accumulation at an earlier age confers longer exposure to the obesity-related metabolic milieu and thus increases the risk for the development of obesity-related morbidity. Therefore the earlier the onset of childhood obesity, the greater is the risk for adult obesity.
Infant overnutrition plays an extremely important role in the future development of obesity. Numerous studies have implicated bottle feeding as a specific risk factor. The prevalence of obesity in children who were never breastfed was 4.5%, as compared with 2.8% in breastfed children, and a clear time-response effect was identified for the duration of breast-feeding on the decline in prevalence of obesity as well. Early overnutrition has been correlated with elevated leptin concentrations in later life. Differences in both volume and composition of commercial formula versus breast milk have been proposed as etiologic factors. An emerging paradigm posits that the gut microbiome plays a critical factor in the development of obesity in childhood as well as in adulthood. Early exposures to maternal factors including breast milk and to other dietary constituents in infancy (such as introduction of solids, exposure to artificial sweeteners etc.) may be key determinants of the profile of the microbiome impacting metabolism and weight balance during childhood and adulthood.
Parental obesity is also an important predictor of childhood obesity. Children with at least one overweight parent at the age of adiposity rebound have a fourfold to fivefold greater chance of becoming adults with obesity. Lean children aged 5 years or younger have a 13-fold risk of adult obesity if both parents have obesity. Excessive BMI gains of parents during childhood and adulthood are also associated with a higher BMI and risk of obesity in the offspring. Conversely, older children with obesity (10–14 years of age) have a 22.3-fold increased risk to become adult with obesity regardless of parental weight, suggesting that parental obesity is more important in early childhood weight gain. Upon studying associations of parental and child obesity status, stronger associations were shown in older children than in younger children , in both parents than in father or mother only, in parental obesity and child obesity compared with overweight status of both. Parental obesity is also related to early adiposity rebound, although it remains unclear whether the relation between parental and childhood obesity is genetic, epigenetic, or environmental.
Metabolic impact of childhood obesity
Many of the metabolic and cardiovascular (CV) complications of obesity are already evident during childhood and are closely related to the development of insulin resistance-hyperinsulinemia, the most common biochemical abnormality seen in obesity. The obesity-related comorbidities that emerge early in childhood are alterations in glucose metabolism, dyslipidemia, and hypertension. Although an accelerated atherogenic process is present in children with obesity, thrombotic CV events do not usually appear until adulthood. The clustering of these manifestations is termed the metabolic syndrome , or the insulin resistance syndrome , suggesting that peripheral insulin resistance may be the driving force of the majority of the obesity-related morbidity.
Insulin Resistance
Insulin resistance is defined as the decreased tissue response to insulin-mediated cellular actions and is the inverse of insulin sensitivity. The term insulin resistance, as generally applied, refers to whole-body reduced glucose uptake in response to physiologic insulin levels and its consequent effects on glucose and insulin metabolism. However, it is now clear that not all insulin-responsive tissues are equally sensitive to insulin. Generalized insulin resistance would result in global metabolic dysfunction, such as leprechaunism or Rabson-Mendenhall syndrome. Thus the insulin resistance of obesity must of necessity affect different tissues quantitatively (see Chapter 3 and Chapter 21 on diabetes mellitus and insulin receptor mutations).
Hepatic Insulin Resistance . The liver plays a major role in substrate metabolism and is the primary target of insulin action. After insulin’s release from the β-cell following a glucose load, it travels directly to the liver via the portal vein, where it binds to the insulin receptor and elicits two key actions at the level of gene transcription. First, insulin stimulates the phosphorylation of FOXO1, which prevents it from entering the nucleus, and thus diminishes the expression of genes required for gluconeogenesis, mainly phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase. The net effect is diminished hepatic glucose production. Second, insulin activates the transcription factor sterol regulatory element-binding protein (SREBP)-1c, which in turn increases the transcription of genes required for fatty acid and triglyceride (TG) biosynthesis, most notably ATP-citrate lyase, acetyl-coenzyme A carboxylase, and fatty acid synthase; which together constitute the process of de novo lipogenesis (DNL). TGs synthesized by DNL are then packaged with apolipoprotein B (apoB) into very low-density lipoproteins (VLDL) for export to the periphery for storage or utilization by reciprocal activation of lipoprotein lipase (LPL) on the surfaces of endothelial cells in adipose or muscle tissues.
For reasons that remain unclear, insulin-resistant subjects typically have “selective” or “dissociated” hepatic insulin resistance; that is, they have impaired glucose homeostasis (mediated by the FOXO1 pathway) but normal insulin-mediated hepatic DNL (mediated by the SREBP-1c pathway and the GCKR gene ). The increase in FFA flux within the liver, either by DNL or FFA delivery via the portal vein, impairs hepatic insulin action via fatty acyl-CoA intermediates within the hepatocyte, leading to increases in hepatic glucose output, the synthesis of proinflammatory cytokines, and excess TG secretion by the liver, low high-density lipoprotein (HDL) cholesterol levels, and an increase of relatively cholesterol-depleted LDL particles. Furthermore, the intrahepatic accumulation of FFA and lipid are also detrimental to liver insulin sensitivity as this leads to the generation of toxic lipid-derived metabolites, such as DAG, fatty acyl CoA, and ceramides. These in turn trigger activation of protein kinase C-ɛ (PKCɛ, and serine/threonine phosphorylation of insulin receptor substrate 1 (IRS-1), which attenuates hepatic insulin signal transduction. Intrahepatic insulin resistance results in greater first pass insulin clearance in the liver, resulting in lower amounts of insulin reaching the systemic circulation.
Adipose Tissue Insulin Resistance . The expanded adipose tissue mass that accompanies obesity often leads to increased lipolysis and FFA turnover. Normally, insulin inhibits adipose tissue lipolysis; however, in the insulin-resistant state, the lipolytic process is accelerated, leading to increased FFA release into the circulation. Moreover, visceral adipocytes are more sensitive to catecholamine-stimulated lipolysis than subcutaneous adipocytes, further increasing FFA flux. Macrophages also infiltrate into adipose tissue and contribute to both adipocyte hypertrophy and cytokine release. These circulating cytokines also affect insulin action in other tissues, such as liver and muscle. Within the normal glucose tolerance range, an increase in adipose insulin resistance is related to an increase in 2-h glucose levels. A tight relation exists between visceral fat (r = 0.34; P < .001) and the visceral/subcutaneous fat ratio and adipose tissue resistance to insulin. Greater FFA concentration following an oral glucose load is also evident with worsening glucose tolerance indicating reduced suppression of lipolysis and lower FFA clearance.
Muscle Insulin Resistance . Downstream of an insulin-resistant liver, increased plasma FFA flux into skeletal muscle results in fatty acyl-CoA derivates altering the insulin signal transduction pathway and resulting in reduced insulin-mediated glucose transport in skeletal muscle, facilitating the development of hyperglycemia. The ectopic deposition in skeletal muscle of fat as intramyocellular lipid may also play a direct role in the pathogenesis of whole-body insulin resistance and metabolic syndrome via lipid metabolite-induced activation of PKCɛ with subsequent impairment of insulin signaling. Greater intramyocellular lipid deposition is tightly associated with insulin resistance and is typically detected in children with obesity who have altered glucose metabolism. The manifestation of impaired insulin signal transduction in skeletal muscle is reduced translocation of GLUT4 to the cell membrane leading to reduced systemic glucose uptake.
Assessment of Insulin Resistance . The euglycemic hyperinsulinemic clamp is the gold standard for measuring insulin sensitivity; the frequently sampled intravenous glucose tolerance test (FSIVGTT) and steady-state plasma glucose (SSPG) methods are also valid measurements. The clamp is performed by infusing a body-surface-area–adjusted continuous insulin drip while maintaining fasting plasma glucose concentrations by modifying a glucose infusion. Greater glucose infusion rates needed to maintain euglycemia indicate greater insulin sensitivity. Euglycemic hyperinsulinemic clamp studies have shown that insulin resistance is determined primarily by the response of skeletal muscle, with over 75% of infused glucose taken up by muscle and only 2% to 3% by adipose tissue. All three methods are generally time consuming, require intravenous infusions and frequent blood sampling, are burdensome for participants, costly, and require a research setting. In an attempt to simplify the measurement of insulin sensitivity, a number of methods using single simultaneously obtained samples of fasting insulin and glucose have been developed, such as the homeostatic model for assessment of insulin resistance (HOMA-IR). Each of these uses a mathematical formula that adjusts for individual variability in insulin and glucose secretion and clearance. Although the goal for these methods was to improve the accuracy of fasting insulin alone by the addition of fasting glucose, it is now agreed that they yield similar results to fasting insulin. When correlated with gold standard methods in children, fasting insulin is a poor measure of whole-body insulin sensitivity in an individual child. Although the primary interest has been in insulin resistance, the adverse effects related to insulin resistance are more likely mediated via compensatory hyperinsulinemia. The fasting triglyceride to HDL-cholesterol ratio, a surrogate of insulin resistance that does not use insulin measurements, has been proposed and correlate quite well with clamp-derived insulin resistance, yet its utilization needs validation in ethnically diverse populations.
The two most important biologic conditions associated with insulin resistance in childhood are ethnicity and puberty. Studies show that African American, Hispanic, Pima Indian, and Asian children are less insulin sensitive compared with non-Hispanic white children with similar anthropometric measures. Insulin resistance in minority ethnic groups is manifested as lower insulin-stimulated glucose uptake, concomitant with hyperinsulinemia, evidence of increased insulin secretion from the β-cell, and decreased insulin clearance. During puberty there is around 25% to 50% decline in insulin sensitivity with recovery when pubertal development is complete. The compensatory increase in insulin secretion during puberty may be blunted in African American and Hispanic youth, thus increasing their risk for type 2 diabetes (T2DM) around the time of puberty. The development of T2DM is covered in depth in Chapter 21 , yet it is worth noting that impaired glucose tolerance (IGT), known as prediabetes, is a relatively common condition in children and adolescents with obesity. IGT in youth with obesity is typically characterized by obesity with an unfavorable pattern of lipid partitioning, with increased deposition of fat in the visceral, hepatic, and intramyocellular compartments.
Lipid Partitioning
The term lipid partitioning refers to the distribution of body fat in various organs and compartments. The majority of excess fat is stored in its conventional subcutaneous depot, yet other potential storage sites exist as well, such as the intraabdominal (visceral) fat compartment and insulin-responsive tissues, such as muscle and liver. One hypothesis to explain the relation between obesity and insulin resistance is the “portal-visceral” paradigm. This hypothesis claims that increased adiposity causes accumulation of fat in the visceral depot, leading to an increased portal and systemic FFA flux ( Fig. 24.7 ). Associations between visceral adiposity, insulin resistance, and comorbidities have been demonstrated across most age groups and ethnicities. Of note, studies of in vivo FFA fluxes from the visceral and the subcutaneous truncal and abdominal depots have failed to demonstrate a substantial difference in net fluxes between these depots.
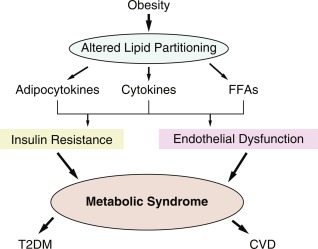
Subcutaneous fat, which does not drain into the portal system, is strongly related to insulin resistance in healthy men with obesity and in men with diabetes. Similarly, truncal subcutaneous fat mass has been demonstrated to independently predict insulin resistance in women with obesity. Visceral and subcutaneous fat differ in their biologic responses because visceral fat is more resistant to insulin and has increased sensitivity to catecholamines. These observations emphasize that both visceral and subcutaneous abdominal fat can contribute to insulin resistance, possibly by different mechanisms. Studies performed in adolescents with obesity highlight the fact that the ratio of visceral to subcutaneous fat may be the determinant of their metabolic impact rather than their absolute quantity of fat. Indeed, adolescents with obesity who have a high visceral to subcutaneous fat ratio, despite having a comparable degree of obesity, demonstrate a markedly adverse metabolic phenotype of severe insulin resistance and alterations in glucose and lipid metabolism. Moreover, intrahepatic fat, although strongly associated with high levels of visceral fat, is also associated with the insulin-resistant state in adolescents with obesity, independent of all other fat depots.
A more unifying paradigm is the “adipose tissue expandability” hypothesis claiming that total body fat is not the culprit of adverse health in obesity rather the relative proportion of lipids in various fat depots is what determines the metabolic phenotype and its derived metabolic risk. This theory claims that as adipose tissue expands, “ectopic lipid deposition” in insulin-responsive tissues is the culprit of adverse effects on metabolism. This theory is based on the observations that lipid content in liver and/or muscle is increased in obesity and in T2DM and is a strong predictor of insulin resistance. Moreover, in conditions such as lipodystrophies, all fat is stored in liver and muscle because of lack of subcutaneous fat tissue, causing severe insulin resistance and diabetes. In adults with obesity (BMI > 30), muscle attenuation on computed tomography ([CT]; representing lipid content) is a stronger predictor of insulin resistance than is visceral fat. Studies performed in vivo using proton nuclear magnetic resonance ( 1 H-NMR) spectroscopy demonstrated increased intramyocellular lipid (IMCL) content to be a strong determinant of insulin resistance in adults and in adolescents with obesity. Alternatively, lipid deposition in hepatocytes to produce intrahepatocellular lipid (IHCL) is highly predictive of insulin resistance, even more so than visceral fat. Thus obesity-driven morbidity may begin when the subcutaneous adipose tissue reaches its capacity to store excess fat and begins to shunt lipid to ectopic tissues, such as liver and muscle, leading to peripheral insulin resistance ; or possibly when liver or muscle accumulates lipid produced de novo in response to dietary manipulation (see later). Another postulated cause of IMCL and IHCL accumulation is a reduction of fat β-oxidation, related to low aerobic capacity, a reduced number or malfunction of mitochondria, or reduced SNS tone. The effect of IMCL or IHCL accumulation on peripheral sensitivity is postulated to be caused by an alteration of the insulin signal transduction pathway in muscle, caused by derivates of fat, such as long-chain fatty acyl-CoA and DAG within the hepatocyte or myocyte. These derivates activate the serine/threonine kinase cascade and cause serine phosphorylation of IRS-1, which inhibits insulin signaling. A comparable mechanism has been demonstrated in the liver, where accumulation of lipids, in particular DAG, activates the inflammatory cascade by inducing c-jun N-terminal kinase 1 (JNK-1), which causes serine rather than tyrosine phosphorylation of IRS-1, leading to inhibition of hepatic insulin signaling.
Vascular Changes
Early stages of the atherosclerotic process may be detected in children with obesity. In recent years, it has become clear that endothelial dysfunction represents a key early step in the development of atherosclerosis. The hallmark and cause of endothelial dysfunction is impairment in nitric oxide (NO)-mediated vasodilatation. This is caused by decreased NO production by endothelial nitric oxide synthase (eNOS), which has been postulated to result from high levels of FFAs and inflammatory cytokines (interleukin [IL]-6, tumor necrosis factor [TNF]-α) in insulin-resistant individuals with obesity, increased reactive oxygen species, or increased uric acid, which inhibit eNOS activity. Decreased NO bioavailability leads to an imbalance between vasodilating and vasoconstricting factors (such as endothelin), which leads to impaired vascular smooth muscle relaxation, increased adhesion of inflammatory cells to the endothelium, increased expression of plasminogen activator inhibitor–1 (PAI-1; a prothrombotic molecule) and increased vascular smooth muscle cell proliferation. Thus decreased NO bioavailability is thought to create a proinflammatory, prothrombotic environment which promotes atherosclerosis. Endothelial function represents an integrated index of the overall CV risk burden in any given individual. During the last decade, noninvasive techniques for the assessment of endothelial function, including high-resolution external vascular ultrasound to measure flow-mediated endothelium-dependent dilatation (FMD) of the brachial artery during hyperemia have been developed. Impaired FMD correlates with arterial wall stiffness, coronary dilatation, and endothelial dysfunction in children with obesity. Similarly, anatomic changes in peripheral arterial vessels, such as increased intimal medial thickness (IMT), have also been demonstrated in children and adolescents with obesity, which mimics early coronary pathology and predicts adverse CV outcomes.
There are no longitudinal studies that directly measure in vivo insulin sensitivity and its relationship to the development of atherosclerotic abnormalities in children. Very limited observations suggest a relationship between HOMA-IR and arterial stiffness and fasting insulin levels in youth. However, a role for insulin resistance in the early abnormalities of vascular smooth muscle is proposed based on the observation that circulating biomarkers of endothelial dysfunction (intercellular adhesion molecule and E-selectin) are highest, whereas adiponectin, the antiatherogenic adipocytokine, is lowest among the most insulin-resistant youth. The landmark Bogalusa heart study demonstrated that CV risk factors present in childhood are predictive of coronary artery disease in adulthood. Among these risk factors, LDL-cholesterol and BMI measured in childhood were found to predict IMT in young adults. There is now substantial evidence that the insulin resistance of childhood obesity creates the metabolic platform for adult CV disease. Obstructive sleep apnea (OSA), typically present in children with obesity, is also tightly associated with the presence of endothelial dysfunction. Moreover, the constellation of peripheral insulin resistance, an unfavorable adipocytokine profile, subacute inflammation, and endothelial dysfunction work in parallel to promote the pathologic processes of aging.
Adipocytokines
Leptin . The discovery of leptin in 1994 has dramatically changed the view of adipose tissue in the regulation of energy balance. Adipocytes secrete several proteins that act as regulators of glucose and lipid homeostasis. These proteins have been collectively referred to as adipocytokines because of their structural similarity with cytokines. Circulating leptin levels correlate with the degree of obesity. As stated earlier, the primary role of leptin is to serve as a long-term energy storage sensor to protect against starvation. Leptin probably has a permissive role in high-energy metabolic processes, such as puberty, ovulation, and pregnancy, but its role in states of energy excess is less known. In obesity, the development of leptin resistance may result in a breakdown of the normal partitioning of surplus lipids in the adipocyte compartment.
Adiponectin. The cytokine adiponectin is peculiar in obesity because, in contrast with the other adipocytokines, its level is reduced in individuals with obesity. The adiponectin gene is expressed exclusively in adipose tissue and codes a protein carboxyl terminal globular head domain and an amino terminal collagen domain, which is structurally reminiscent of the complement factor 1q. The gene is located on chromosome 3q27, a location previously linked to the development of type 2 diabetes and the metabolic syndrome. Several single nucleotide polymorphisms (SNPs) in the adiponectin gene have been reported to be associated with the development of type 2 diabetes in populations around the world, suggesting that adiponectin plays a major role in glucose metabolism. Adiponectin circulates in plasma in three major forms: a low-molecular-weight trimer, a middle-molecular-weight hexamer, and a high-molecular-weight 12- to 18-mer. Circulating plasma high-molecular-weight adiponectin concentrations demonstrate a sexual dimorphism (females have greater concentrations), suggesting a role for sex hormones in the regulation of adiponectin production or clearance. Dietary factors, such as linoleic acid or fish oil versus a high carbohydrate diet or increased oxidative stress, have been shown to increase or decrease adiponectin concentrations, respectively. These observations suggest that the circulating levels of adiponectin are regulated by complex interactions between genetic and environmental factors.
The receptors for adiponectin have been characterized in rodent models and cloned. Two receptors, named ADIPOR1 and ADIPOR2, have been characterized. ADIPOR1 is expressed in numerous tissues including muscle, whereas ADIPOR2 is mostly restricted to the liver. Both receptors are bound to the cell membrane, yet are unique in comparison to other G-protein–coupled receptors in the fact that the C-terminal is external, whereas the N-terminal is intracellular. Both ADIPOR1 and ADIPOR2 are receptors for the globular head of adiponectin and serve as initiators of signal transduction pathways that lead to increased PPARα and increased adenosine monophosphate (AMP) kinase activities, which promote glucose uptake and increased fatty acid oxidation. Adiponectin has been shown to have potent antiatherogenic functions, as it accumulates in the subendothelial space of injured vascular walls to reduce the expression of adhesion molecules and the recruitment of macrophages.
Studies in children and adolescents with obesity have shown that adiponectin is inversely related with the degree of obesity, insulin sensitivity visceral adiposity, IHCL, and IMCL, whereas weight loss increases adiponectin. In adolescents with obesity and type 2 diabetes, low baseline adiponectin and a reduced elevation in response to treatment have been shown to predict treatment failure. All of these observations along with human clinical data support a pivotal role for adiponectin in the prevention of the comorbidities of the metabolic syndrome.
Family studies using parent-offspring regressions revealed that most adipocytokines show evidence for significant inheritance. There are three main common axes of variation in the heritability of adipocytokines. The main axis, which explained 21% of the variation, was most strongly loaded on levels of leptin, TNF-α, insulin, and PAI-1, and inversely with adiponectin. This axis was significantly associated with BMI and phenotypically stronger in children, and showed a heritability of 50%, after adjustment for age, gender, and generational effects. Thus adipocytokines are highly heritable and their pattern of covariation is significantly correlated with BMI as early as the preteen years.
Myokines and Natriuretic Peptides. Skeletal and heart muscles may serve as an endocrine organ as well. Some of the effects of exercise on skeletal muscle are mediated by the transcriptional coactivator PPAR-γ coactivator 1α (PGC-1α). In the mouse, PGC-1α expression in muscle stimulates an increase in expression of FNDC5, a membrane protein that is cleaved and secreted as a newly identified hormone, named Irisin . Irisin acts on white adipose cells in culture and in vivo to stimulate UCP1 expression and induces “beiging” of white adipocytes into cells metabolically more active. Irisin is induced with exercise in mice and humans, and mildly increased irisin levels in the blood cause an increase in energy expenditure in mice with no changes in movement or food intake, resulting in improvements in obesity and glucose homeostasis. This novel myokine is actually the first hormonal link between exercise and the adipose tissue changes it may induce. Importantly, this molecule has a negative correlation with brain executive function and thus probably has multiple effects yet to be discovered. Atrial natriuretic peptides (ANP) also have been implicated in fat metabolism. These natriuretic peptides are produced with exercise, cardiac wall stress, weight loss, and cold exposure, and inhibited by obesity and insulin resistance. ANP binds to its natriuretic receptor, facilitating the formation of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP). cGMP phosphorylates cGMP-dependent protein kinase, which activates lipolysis, and phosphorylates p38 mitogen-activated protein kinase to enhance mitochondrial biogenesis with increased energy expenditure and increased heat generation, as part of the brown-fat thermogenic program. Thus both skeletal and cardiac muscle may respond to exercise by inducing changes in fat metabolism to enhance caloric expenditure and limit obesity. These new findings are likely to open new avenues of clinical research to limit the consequences of the “obesity epidemic.”
Inflammatory Cytokines. Accumulating evidence indicates that obesity is associated with subclinical chronic inflammation. Adipose tissue serves not merely as a simple reservoir of energy stored as TGs, but also as an active secretory organ releasing many peptides, including inflammatory cytokines, into the circulation. This is probably because of infiltration of adipose tissue by cells of the immune system, mainly macrophages. In obesity, the balance between these numerous peptides is altered, such that larger adipocytes and macrophages embedded within them produce more inflammatory cytokines (i.e., TNF-α, IL-6) and less antiinflammatory peptides, such as adiponectin. One hypothesis posits that as energy accumulates in adipocytes, the perilipin border of the fat vacuole breaks down, causing the adipocyte death. Cell death recruits macrophages into the adipose tissue, especially within the visceral compartment, which in the process of clearing debris, also elaborate inflammatory cytokines, initiating a proinflammatory milieu that predates and possibly drives the development of systemic insulin resistance, diabetes, and endothelial dysfunction. Systemic concentrations of C-reactive protein (CRP) and IL-6, two major markers and participants of the inflammatory process, are increased in children and adolescents with obesity. CRP levels within the “high-normal” range have been shown to predict CV disease and development of T2DM in adults. Elevated levels of CRP correlate with other components of the metabolic syndrome in children with obesity. Thus inflammation may be one of the links between obesity and insulin resistance, potentially driving myocyte and hepatic resistance within the insulin signal transduction pathway.
Reactive Oxygen Species (ROS). The “Free Radical Theory” holds that imbalance between ROS generation and antioxidant defenses is a major factor in the determination of lipid peroxidation and protein misfolding, with resultant deoxyribonucleic acid (DNA) and cellular damage. Excessive intracellular ROS formation occurs via three pathways: (1) inflammatory cytokines derived from visceral fat accumulation, (2) dysfunctional mitochondrial energetics, and (3) glycation. Excessive nutrient processing by mitochondria can result in uncoupling of oxidative phosphorylation and increased generation of ROS; this, in turn, leads to altered mitochondrial function and further ROS generation. ROS accumulation can also impair ER function, causing ER stress and the compensatory unfolded protein response (UPR). The UPR can itself be overwhelmed by persistent excessive nutrient processing and ROS generation, leading to cellular shutdown, defective insulin secretion, and T2DM ( Fig. 24.8 ).
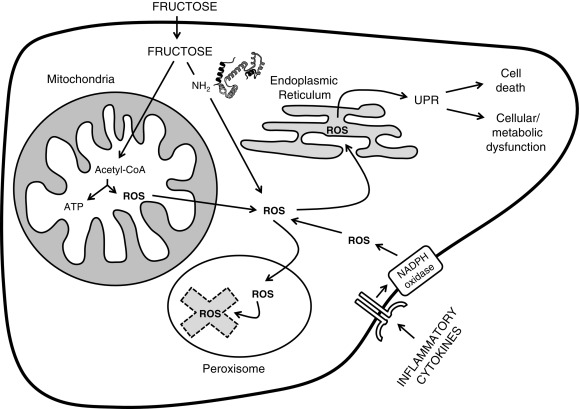
Because ROSs are inherent by-products of cellular metabolism, endogenous cellular antioxidants (e.g., catalase and glutathione) quench the ROS before they have a chance to promote peroxidation. These antioxidants are found primarily in peroxisomes, which collaborate with the mitochondria in ROS processing. Reduction in peroxisomal activity results in mitochondrial dysfunction and ER stress. Furthermore, cytokines, such as TNF-α, can reduce peroxisomal number and function, rendering cells even more vulnerable.
Comorbidities Related to Insulin Resistance
The Metabolic Syndrome
The association and clustering of T2DM, hypertension, dyslipidemia, and CV disease in adults has led to the hypothesis that they may arise from a common antecedent. The WHO argues that this antecedent is insulin resistance, and defines this association as the metabolic syndrome. A consensus definition of the metabolic syndrome for the pediatric age group has been published and declares that children younger than 10 years of age should not be defined as having this condition. For children older than 10 years of age, the obesity component of the definition is waist circumference and not BMI, indicating the clinical importance of intraabdominal fat. The metabolic syndrome affects approximately 25% of the US adult population. Because of its wide prevalence, the metabolic syndrome is of enormous clinical and public health importance, even at its earliest stages. Although still debated, one scheme of the pathophysiology of the metabolic syndrome is shown in Fig. 24.7 . According to this paradigm, the impact of obesity is determined by the pattern of lipid partitioning—that is, the specific depots in which excess fat is stored. This pattern of lipid storage determines the adipocytokine secretion profile, on circulating concentrations of inflammatory cytokines and on the flux of FFA. The combined effect of these factors determines the sensitivity of insulin target organs (such as muscle and liver) to insulin and impacts the vascular system by affecting endothelial function. Peripheral insulin resistance and endothelial dysfunction are the early promoters of overt pathology, culminating in T2DM and CV disease. Regardless of the metabolic syndrome definition used, insulin resistance and high insulin levels are associated with the clustering of cardiometabolic risks associated with metabolic syndrome in a variety of ethnic groups. It should be noted that when studying a population (not necessarily an individual), increasing degrees of obesity in childhood are associated with greater risk for the presence of CV risk factors, yet this risk seems to plateau above a threshold of obesity which corresponds to 40 kg/m 2 in adults.
Nonalcoholic Fatty Liver Disease
Nonalcoholic fatty liver disease (NAFLD) represents fatty infiltration of the liver in the absence of alcohol consumption. The spectrum of NAFLD ranges from pure fatty infiltration (steatosis) to inflammation (nonalcoholic steatohepatitis, or NASH), to fibrosis and even cirrhosis. NAFLD was found in the NHANES III survey to be more prevalent in African American and Hispanic males with obesity, T2DM, hypertension, and hyperlipidemia. These associations have led to the hypothesis that NAFLD is an early marker of the presence of insulin resistance and appears before the development of overt diabetes. NAFLD is now the most common liver disease among children in North America. NAFLD in children is associated with increased visceral fat deposition, and may progress to cirrhosis and related complications. The association between abdominal obesity and fatty liver may be partially explained by sustained exposure of the liver to an increased flux of FFA from the visceral depot. NAFLD may represent an early manifestation of ectopic lipid deposition in the liver and represents a challenge to the clinician because of the contrast of its minimal early manifestations and its potential serious outcomes. Studies using the hyperinsulinemic euglycemic clamp methodology demonstrate that NAFLD is associated with hepatic and peripheral insulin resistance.
Insulin plays a key role in regulating transcription factors, such as SREBP-1c, which are abundantly expressed in the liver. SREBP-1c is pivotal in the control of hepatic lipogenesis and is increased in proportion to circulating insulin levels. These data raise the possibility that fasting hyperinsulinemia may contribute to hepatic steatosis, rather than vice versa. Alternatively, inflammatory cytokines released by visceral fat or by the hepatic immune-reactive cells may contribute to altered hepatic lipid metabolism. It has been shown that specific SNPs, such as the rs58542926 SNP in the TM6SF2 gene, are associated with pediatric NAFLD, explaining why some but not all children with obesity develop this phenotype. The majority of patients probably experience NAFLD without progressing on to NASH. It is likely that subsequent inflammation from increased ROS formation without appropriate quenching is necessary to promote progression to NASH (the so-called second hit theory). For the time being, NAFLD can be surmised by an elevated alanine aminotransferase (ALT) in children with obesity. However, ALT does not have to be very elevated; the 95 th percentile for ALT in children is 25.8 U/mL for boys and 22.1 U/mL for girls. Importantly, a normal ALT concentration does not rule out the presence of NAFLD.
Polycystic Ovarian Syndrome
The association of hyperandrogenism and oligomenorrhea or amenorrhea in females, termed polycystic ovarian syndrome ( PCOS) , is a frequent comorbidity of obesity, which can extend down to childhood. This disorder is covered in detail in Chapter 16 . The diagnosis of PCOS must be based on the presence of at least two of the following three criteria: chronic anovulation, hyperandrogenism (clinical or biologic), and polycystic ovaries. PCOS is the most common cause of infertility because of anovulation and a major risk factor for development of the metabolic syndrome and altered glucose metabolism in females. The antecedents of PCOS have been identified in prepubertal girls, suggesting a developmental lesion.
Adolescent girls with PCOS can have moderate to severe insulin resistance with increased risk for altered glucose metabolism and the impairment in insulin sensitivity is more pronounced in those with obesity compared with girls with PCOS who were lean. Typically, girls with PCOS and obesity demonstrate altered lipid partitioning including high visceral and hepatic lipid content, as well as decreased lipid mobilization, diminished fat oxidation, and metabolic inflexibility. In some ethnic groups, girls with premature pubarche, a potential antecedent of PCOS, have relatively increased insulin levels, thus a causal link between hyperinsulinemia and androgen hypersecretion (of adrenal or ovarian origin) has been hypothesized. Population studies of normal girls have shown that rapid weight gain is associated with higher adrenal androgens and body fatness, and that hyperinsulinemia is related to early menarche. Thus the association of higher insulin levels with premature pubarche and subsequent PCOS may be driven, at least in part, by obesity. Obesity characterizes about 50% of women with classic PCOS, although it is even more common among adolescents. Increased peripheral insulin resistance occurs in approximately 50% of patients with PCOS, and almost certainly plays a role in the pathogenesis of this condition. On the other hand, almost all forms of severe insulin resistance, such as T2DM or rare lipodystrophy syndromes, are also associated with PCOS. Of note, insulin-resistance has not been included as a diagnostic criterion for PCOS mainly because of the difficulty of its measurement. Fasting hyperinsulinemia and an increased insulin secretory response to an oral glucose load have been demonstrated in girls with PCOS. Indeed, adolescent girls with PCOS and obesity have been shown to be 50% more insulin resistant than weight-matched controls without PCOS. The constellation of metabolic abnormalities typically seen in insulin resistant individuals is commonly encountered in adolescents with PCOS and obesity, including NAFLD and T2DM. The increased prevalence of the metabolic syndrome may be related to the hyperandrogenism independent of obesity-related insulin resistance. Early markers of accelerated atherogenesis are already present in young females with PCOS, indicating that early intervention aimed at reducing CV risk may be beneficial. Importantly, weight loss in women with PCOS is associated with improved menstrual function.
Metabolic examination of patients with PCOS demonstrates hepatic and muscle resistance, but not ovarian insulin resistance; possibly accounting for insulin stimulation of theca cell androgen production. The correlation between insulin resistance and hyperandrogenism begs a unifying hypothesis as to their pathogenesis, which is proffered by the “serine phosphorylation hypothesis,” which suggests that both P450c17 and the insulin receptor are aberrantly serine phosphorylated; in the case of P450c17, this leads to excess activity and increased androgen production, and in the case of the insulin receptor, this leads to tissue-specific insulin resistance. However, this hypothesis remains to be proven.
Other Endocrine Comorbidities
Obesity causes changes in other hormonal systems, some of which confer specific morbidities. The age of pubertal initiation has been creeping earlier, particularly in African American girls. This advancement is explained in part by the increasing overnutrition and BMI seen in this population. Infertility in older adolescents and adult women may occur either as a manifestation of PCOS because of excessive ovarian androgen production in females, or because of excessive aromatization of androgen to estrogen by peripheral adipose tissue with suppression of the hypothalamic-pituitary gonadal axis in both sexes. The hyperestrogenemia may also promote gynecomastia in males. In addition, the hypercapnia associated with OSA can suppress hypothalamic gonadotropin hormone (GnRH) function, leading to a syndrome of delayed puberty.
Obesity is associated with decreased GH secretion, and indeed most subjects with obesity, despite normal or excessive statural growth, fail GH stimulation testing. However, caloric restriction for 24 hours can restore normal GH responsivity. Despite the functional GH inadequacy, statural growth is accelerated, bone age is advanced, and peripheral total and free insulin-like growth factor (IGF)-1 levels are normal or elevated in obesity, suggesting normal or accentuated GH sensitivity, or possibly because of the suppression of insulin-like growth factor-binding protein (IGFBP)-1, and the effects of hyperinsulinemia on activation of the growth plate IGF-1 receptor. Free thyroxine levels tend to be lower and TSH higher in children with obesity, although mostly within the normal range along with some TSH levels within the subclinical hypothyroidism category; the mechanism is unknown. The elevated TSH levels in obesity seem a consequence rather than a cause of obesity. Therefore treatment of hyperthyrotropinemia with thyroxine seems unnecessary in children with obesity. Lastly, obesity can be associated with increased cortisol exposure, possibly because of conversion of circulating cortisone to cortisol by the enzyme 11-β-hydroxysteroid dehydrogenase type 1 (11β HSD1) located within adipocytes.
Other Nonendocrine Comorbidities
Childhood obesity is associated with numerous other comorbidities. Pseudotumor cerebri is a rare and poorly understood condition leading to intracranial hypertension, whose manifestations include papilledema and headache. Treatment includes serial lumbar punctures, acetazolamide to reduce CSF production, in severe cases there is a need for a ventriculoperitoneal shunt, and occasionally optic nerve sheath fenestration is necessary to save eyesight. OSA occurs frequently in children with severe obesity; presumably because of the large amount of retropharyngeal fat that compresses the upper airway during sleep. Affected patients snore, often stop breathing for more than 20 seconds during sleep, and wake up during the night with headache. Treatment includes nocturnal positive airway pressure and, when appropriate, tonsilloadenoidectomy; however, symptoms often recur. Children with obesity manifest numerous orthopedic difficulties, including fractures, knee pain, anatomic lower limb malalignment, and impairment in mobility. Cholelithiasis occurs in approximately 2.5% of adolescents with obesity, especially in females, but is not usually seen in prepubertal children with obesity. Lastly, psychologic distress, including clinical depression, is clearly manifested in children with obesity and specifically in those with severe obesity. These various comorbidities all appear to be associated with BMI z-score in a curvilinear fashion ; thus the more their obesity, the more likely patients will manifest comorbidity.
Factors associated with the current epidemic of obesity
Genetics
The association between obesity and genetics owes to two separate lines of investigation: (1) the discoveries of monogenic disorders of the energy balance pathway (see later) and (2) studies of specific racial and ethnic groups, in which obesity seems to segregate, such as the Pima Indians and Hispanics in the Southwest United States. These observations are combined with an attractive theory on the natural selection of individuals in response to drastic environmental/ecologic pressure (i.e., famine), termed the thrifty gene hypothesis , to yield a very strong driving force for the elucidation of specific genetic loci in the pathogenesis of obesity. However, the rapid timescale of increased prevalence of childhood obesity cannot possibly reflect a population genetic change. Therefore the current model is that obesity is a result of gene-environment interactions; an ancient genetic selection to deposit fat efficiently may have provided a survival advantage in earlier times but is maladaptive with our current food overabundance. An evolutionary approach to obesity development involves a genomic/anthropologic dimension. For millions of years, the lifestyle of hunter-gatherers comprised intense physical activity and a high-protein/low-carbohydrate diet and the genomes of our ancestors were adapted to low insulin sensitivity. Upon development of farming, a diet high in carbohydrates emerged and since the industrial revolution, our genome is rapidly adapting to such diet. Our current genome represents a mixture of the two. Upon exposure to energy excess induced by the current food environment, these genomes may manifest as development of obesity at different timing and may respond differently to specific diets.
In the common forms of obesity, relating SNPs with associated risks for obesity is difficult because the effects are uncertain, and the results not always confirmed. Several SNPs in specific genes have been identified, such as the fat mass and obesity associated ( FTO ) gene. Variation in the FTO gene has provided the most robust associations with common obesity to date yet the FTO variant that confers a predisposition to obesity does not appear to be involved in the regulation of energy expenditure but may have a role in the control of food intake and food choice, suggesting a link to a hyperphagic phenotype or a preference for energy-dense foods. The large genome-wide association studies (GWAS) have yielded to date more than 300 loci that may be relevant to the development of adult obesity, yet explain less than 5% of the phenotype.
Epigenetics and Developmental Programming
Follow-up studies of newborns born small-for-gestational age (SGA), large-for-gestational age (LGA), and premature, have noted markedly increased risks for obesity and the metabolic syndrome. The “fetal origins hypothesis” states that some aspect of the in utero environment contributes to the development of obesity and chronic disease in later life. Importantly, each of these three antenatal conditions is associated with insulin resistance. The “developmental model” of chronic diseases postulates that early-life events affect individual differences in vulnerability to lifestyle and environment. One possible mechanism for such developmental programming includes epigenetic changes, which may contribute to alterations in gene expression. For instance, the pattern of DNA methylation noted in cord blood predicts the degree of adiposity at age 9 years.
Documentation of the relationship of SGA with adult obesity and CV disease started with studies of the Dutch famine during World War II and its aftermath. Several studies of newborns born SGA demonstrate that they are hyperinsulinemic and insulin resistant at birth, exhibit rapid catch-up growth in the early postnatal period, and develop obesity in childhood, which remains and promotes persistent insulin resistance. An analysis of Indian newborns born in India versus the United Kingdom demonstrated that despite those born in India weighing 700 g less at birth, their glucose and insulin levels are markedly elevated. After adjustment for birth weight, the India-born babies demonstrate increased adiposity, 4 times higher insulin, 2 times higher leptin levels than the UK-born babies. Thus these babies are insulin resistant even at birth, which translates into increased adiposity. Following such babies into childhood, there are numerous studies documenting insulin resistance during early childhood.
Babies born LGA are often hyperinsulinemic at birth. Although most LGA babies are caused by gestational diabetes mellitus (GDM) and exposure to maternal obesity and hyperglycemia throughout the pregnancy, this is not always the cause. Follow-up of LGA babies without GDM demonstrates a doubling of prevalence of insulin resistance and metabolic syndrome, whereas LGA babies resulting from GDM manifest a threefold increase. It has been shown that adolescents with obesity who were exposed in utero to GDM are more insulin resistant and have impaired β-cell function compared with peers with equal degree of obesity without such exposure. Indeed, the “vertical” transmission of maternal diabetes to the offspring in the form of later obesity and diabetes has been documented in studies of Pima Indians. Lastly, weight gain during pregnancy increases birth weight, the risk for LGA, and obesity in the offspring ( Fig. 24.9 ).
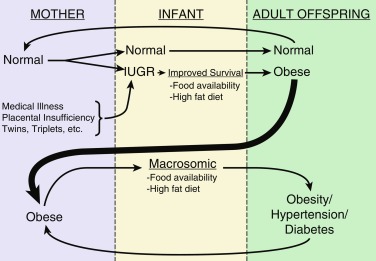
The protective effect of breastfeeding against development of future obesity has long been known, and there appears to be a dose–response; the longer the breastfeeding, the more protective. However, this may be complicated by confounding factors, such as socioeconomic status, maternal smoking in pregnancy, and maternal BMI. The mechanism of breastfeeding’s antiobesity effect is also unclear. Some think infant feeding self-regulation is most relevant, whereas a recent study suggests that leptin in breast milk may contribute to this protection, and finally, maternal breastfeeding may shape the offspring intestinal microbiome into a profile different than that induced by consumption of formula.
Environmental Factors
Numerous environmental factors have also been associated with the obesity epidemic, particularly in children. However, most of these associations are derived from cross-sectional rather than longitudinal studies, and in many instances, mechanism remains lacking. Several longitudinal studies in adults have clearly demonstrated that specific dietary and other lifestyle behaviors are independently associated with long-term weight gain, with a substantial aggregate effect. For instance, on the basis of increased daily servings of individual dietary components, 4-year weight change was most strongly associated with the intake of potato chips (0.767 kg), potatoes (0.58 kg), sugar-sweetened beverages (0.45 kg), unprocessed red meats (0.43 kg), and processed meats (0.42 kg) and was inversely associated with the intake of vegetables (-0.1 kg), whole grains (-0.168 kg), fruits (-0.224 kg), nuts (-0.25 kg), and yogurt (-0.37 kg) ( P ≤ .005 for each comparison). Similarly, the sociodemographic environment has been shown to affect the chance of having obesity in adulthood. Indeed, the opportunity to move from a neighborhood with a high prevalence of poverty to one with lesser poverty was associated with modest but potentially important reductions in the prevalence of extreme obesity and diabetes. Similar relationships are likely, but not proven, for children and adolescents.
Stress and Cortisol
In humans, elevated cortisol or markers of the hypothalamus, pituitary, and adrenal (HPA) axis dysregulation correlate with abdominal fat distribution and the metabolic syndrome. Although circulating cortisol is clearly important in determining visceral adiposity, the recent identification of reduction of circulating cortisone to cortisol within visceral fat tissue by the enzyme 11βHSD1 has also been linked to the metabolic syndrome. These data suggest that cortisol is important both in increasing visceral adiposity and promoting the metabolic syndrome. The mechanistic link between stress and obesity has not been clarified, partly because of the inherent complexity of evaluating a potentially bidirectional effect of stress on eating and body weight. Studies focusing on brown adipose tissue metabolism support a dichotomous relation to explain the impact of stress on obesity: stress promotes obesity in the presence of hyperphagia and stable brown adipose function, whereas stress results in weight loss or protection from obesity development in the presence of hypophagia or when increased calorie intake is associated with brown adipose recruitment and enhanced thermogenesis (beiging of white adipocytes).
Evidence of associations between elevated cortisol and psychologic distress with abdominal fat distribution in adults is compelling. For instance, urinary glucocorticoid excretion is linked to aspects of the metabolic syndrome, including blood pressure, fasting glucose, insulin, and waist circumference. It seems that some individuals seem to be “high-responders” to a stress stimulus and demonstrate higher cortisol secretion. These individuals seem more prone for an alteration in satiety recognition and consume larger amounts of calories following the stress exposure (see Fig. 24.5 ). These data suggest that cortisol is important both in increasing visceral adiposity and promoting insulin resistance, equivalent to “Cushing syndrome of the abdomen.”
Sleep Deprivation
Adults in the United States currently average less than 7 hours of sleep per night—almost 2 hours less than in 1980—and about one-third of them get less than 6 hours per night. Analyses of data from the NHANES I, revealed that adults (ages 32–49 years) who got less than 7 hours of sleep were more likely to have obesity 5 to 8 years later than those who got 7 or more hours of sleep. The link between short sleep duration and obesity has also been observed among children. Longitudinal studies in children, with subjects from diverse backgrounds, suggested an inverse association between sleep duration and BMI. In 24,821 participants, pediatric subjects sleeping for short duration had twice the risk of having overweight/obesity, compared with subjects sleeping for long duration (odds ratio 2.15; 95% CI, 1.64-2.81). Like adults, increasing numbers of children are chronically sleep deprived. This is especially true of children with obesity, who have been found to get less sleep than those of normal weight. In addition to its other effects, sleep is one of the most powerful cross-sectional and longitudinal predictors of childhood obesity in prepubertal children.
Television Viewing and “Media Time”
Television watching is considered one of the most modifiable causes of childhood obesity. There are four possible mechanisms linking television watching and obesity. Firstly, television watching may increase stress levels and cortisol (see earlier), causing increased food intake, and promotion of obesity. Second, television watching displaces physical activity. Most, but not all studies find inverse correlations between television watching and physical activity and fitness. Third, television watching increases calorie consumption from eating during viewing or from the effects of food advertising. Television viewing is also associated with increased high-fat food intake, decreased fruit and vegetable consumption, and increased soft drink intake. “Junk food” is the most frequently advertised product category on children’s television. Lastly, REE and NEAT appear to be decreased during television watching. According to NHANES III, the prevalence of childhood obesity is lowest among children watching television for 1 hour or less a day, and highest among those watching 4 hour or more a day. Several experimental studies of reducing television watching have been conducted and their results support the suggestion that reduced television watching may help to reduce the obesity risk or help promote weight loss in children with obesity. These studies represent the strongest direct evidence that altering television watching alone is a promising strategy for prevention of childhood obesity. Other forms of screen time, such as cell phones, are also implicated in obesity pathogenesis. It has been shown that parental monitoring of child media exposure predicted lower child BMI z-scores at age 7 years and less steeply increasing child BMI z-scores from 5 to 9 years, thus parental behaviors related to children’s media consumption may have long-term effects on children’s BMI in middle childhood.
Dietary Factors
Calories from any food have the potential to increase risk for obesity and cardiometabolic disease because all calories can directly contribute to positive energy balance and fat gain. However, various dietary components may promote obesity and cardiometabolic disease by additional mechanisms that are not mediated solely by their caloric content. Regarding the health effects of specific dietary elements, it has been shown that food-specific saturated fatty acids and sugar-sweetened beverages promote cardiometabolic diseases by mechanisms that are additional to their contribution of calories to positive energy balance. Metabolic effects and responses to certain dietary components are influenced by the individual’s metabolic status, developmental period, or genotype; by the responsiveness of brain regions associated with reward to food cues; and possibly by the microbiome.
Dietary Fat Versus Carbohydrate
Fat is generally considered more obesogenic than other macronutrients, because it is more energy dense, highly palatable, and more effectively converted to body fat. A high-fat meal induces less thermogenesis and a higher positive fat balance than an isocaloric and isoprotein containing low-fat meal. Excessive fat intake is believed to cause weight gain, but the relationships between dietary fat intake and childhood adiposity remain controversial.
The prevalence of overweight in the United States has increased despite a decreased percentage of dietary energy derived from fat. A metaanalysis of 12 studies in adults with overweight or obesity adults who were given dietary advice on low-fat diet and followed for 6 to 18 months suggested that low-fat diets are no better than calorie-restricted diets in long-term weight loss. Similarly, in children, total fat consumption expressed as a percentage of energy intake has decreased. This decrease in fat consumption is largely caused by increased total energy intake in the form of carbohydrates. Much of this imbalance is attributed to changing beverage consumption patterns, characterized by declining milk intakes and substantial increases in soft-drink consumption, which may have its own etiopathogenesis (see later). Most interventions with a low-fat, heart-healthy diet, have not been successful in childhood overweight prevention.
Reduction in carbohydrate intake is taken to the extreme in the ketogenic diets, such as the Atkins diet, which restricts adult subjects to less than 25 g/d of ingested carbohydrate. Adult evaluations of the diet have been disappointing long term, and the popular diet has been abandoned recently. There are currently no data in children or adolescents. However, it should be noted that the ketogenic diet used for seizure control is similar in composition to the Atkins diet. A 2-year study of the ketogenic diet demonstrated persistent decreases in weight z-scores in children who were above average upon diet initiation, without significant compromise in general nutrition or in height.
Trans Unsaturated Fatty Acids (Trans-Fats)
Trans -unsaturated fats in processed foods have been a staple in the Western diet since the early 20 th century. This is because the trans-isomerization of the double bond prevents fatty acid breakdown by bacteria, prolonging the shelf life of food products. Like its bacterial predecessors, human mitochondria cannot subject trans -fats to β-oxidation in the liver, contributing to ectopic intrahepatic lipid accumulation. Fortunately, because of the recognized association between trans -fat consumption and CV disease in the mid-1980s and more stringent labeling requirements since 2006, the percent of calories from trans -fats consumed in the Western diet has been gradually declining. Trans -fats have no health benefit and cause hepatic steatosis and insulin resistance ; however, their current consumption trends are temporally disparate with the current increasing prevalence of metabolic syndrome, suggesting that other factors are involved.
Glycemic Index and Fiber
Not all sugars exert the same insulinogenic response. Complex carbohydrates can take two forms: either a combination of α1-4 linkages and α1-6 linkages, which gives the starch a globular structure called amylopectin , as seen in bread, rice, pasta, potatoes, and glycogen; or a linear polymer of α1-4 linkages called amylose , as seen in beans, lentils, and other legumes. Digestion and absorption of the former in the intestine is rapid because of the simultaneous actions of both α1-4 and α1-6 glucosidases, whereas that of the latter is much slower because the α1-4 glucosidase can only cleave single glucose moieties on either side of the polymer. This phenomenon constitutes the basis of the glycemic index (GIx), which refers to the relative glucose area under the curve after consumption (in comparison with dextrose). High-GIx foods lead to an accentuated insulin response, which can shunt energy substrate to adipose tissue. In children, controlled studies with a high-GIx diet demonstrate that energy intake is 53% higher than on low-GIx diet. One adolescent study demonstrated that an ad libitum low-GIx diet was more effective in promoting weight loss than an energy-restricted low-fat diet. Therefore the GIx may be a simple concept to institute, although the “toxic environment” of American food products may make it difficult to maintain.
Dietary fiber consists of the nonstarch, polysaccharide portion of plant foods, including cellulose, hemicellulose, pectins, β-glucans, fructans, gums, and algal polysaccharides. Major sources of dietary fiber include whole grains, fruits, vegetables, legumes, and nuts. Fiber content accounts for 50% of the variability in glycemic load (GL; GIx × volume) between foods. Cohort studies of adults demonstrate that fiber intake is inversely associated with weight gain, fasting insulin levels, and risk of T2DM. Fiber may influence body weight regulation by several mechanisms involving intrinsic, hormonal, and colonic effects, which eventually decrease food intake by promoting satiation (lower meal energy content), satiety (longer duration between meals), or by increasing fat oxidation and decreasing fat storage. A fiber-rich meal is processed more slowly and has less caloric density and lower in fat and added sugars. Fiber-containing foods engender slower glucose absorption, which lessens the postprandial insulin surge and decreases lipogenesis. In addition, high-fiber meals allow for delivery of undigested triglyceride to the colon, where fermentation to short-chain fatty acids and their absorption improve lipids and insulin sensitivity. Archeologists surmise that our ancestors consumed 100 to 300 g of fiber/d. However, the dietary fiber intake throughout childhood and adolescence currently averages approximately 12 g/d, and has not changed during the past 30 years. Therefore parents and school foodservice personnel should strive to offer fiber-rich foods to children so their acceptance and consumption of them will be increased.
Fructose
The most commonly used sweetener in the US diet is the disaccharide sucrose (e.g., table sugar), which contains 50% fructose and 50% glucose. However, in North America and many other countries, nondiet soft drinks are sweetened with high-fructose corn syrup (HFCS), which contains up to 55% of the monosaccharide fructose. Thanks to its abundance, sweetness, and low price, HFCS has become the most common sweetener used in processed foods. It is not that HFCS is biologically more ominous than sucrose; rather, it is that its low cost has made it available to everyone, especially low socioeconomic groups. HFCS is found in processed foods ranging from soft drinks and candy bars to crackers to hot dog buns to ketchup. Average daily fructose consumption has increased by over 25% over the past 30 years. The growing dependence on fructose in the Western diet may be fueling the obesity and T2DM epidemics. The highest fructose loads are soda (1.7 g/30 mL) and juice (1.8 g/30 mL). Although soda has received most of the attention, high fruit juice intake is also associated with childhood obesity, especially by lower income families. Animal models demonstrate that high-fructose diets lead to increased energy intake, decreased resting energy expenditure, excess fat deposition, and insulin resistance, which suggest that fructose consumption is playing a role in the epidemics of insulin resistance and obesity and T2DM in humans.
Fructose in the gut is transported into the enterocyte via the fructose transporter GLUT5, independent of ATP hydrolysis and sodium absorption. Once inside in the enterocyte, a small portion of the fructose load is converted to lactic acid and released in the portal circulation, another small portion may also be converted to glucose. However, the majority of ingested fructose is secreted into the portal circulation and delivered to the liver. There, fructose is rapidly metabolized to fructose-1-phosphate (F1P) via fructokinase, an insulin-independent process, which also bypasses the negative feedback regulation of phosphofructokinase in the glycolytic pathway. Thus fructose metabolism generates lipogenic substrates (e.g., glyceraldehyde-3-phosphate and acetyl-CoA) in an unregulated fashion, which are delivered directly into the mitochondria. This excessive mitochondrial substrate then drives hepatic DNL, leading to intrahepatic lipid deposition and steatosis. Hepatic DNL also limits further fatty acid oxidation in the liver via excess production of malonyl-CoA, which reduces entry of fatty acids into the mitochondria by inhibiting carnitine palmitoyl transferase 1 (CPT-1). F1P also stimulates SREBP-1c via peroxisome proliferator-activated receptor gamma coactivator (PGC)-1β, independently of insulin, which activates the genes involved in DNL; moreover, fructose has been shown to induce activation of carbohydrate-response element binding protein (ChREBP), which also increases the expression of all the enzymes of DNL. Furthermore, F1P activates dual-specificity mitogen-activated protein kinase 7 (MKK7), which subsequently stimulates Janus kinase 1 (JAK1), a hepatic enzyme considered to act as a bridge between hepatic metabolism and inflammation. In addition, the lipogenic intermediate DAG (formed during fructose metabolism in the liver) activates PKCɛ, which phosphorylates serine residues on IRS-1, inactivating it, and leading to hepatic insulin resistance. This impairs insulin-mediated phosphorylation of FOXO1, leading to increased expression of the genes required for gluconeogenesis and promoting increased hepatic glucose output, also contributing to hyperglycemia and the development of T2DM. The excess TGs secreted from the liver into the circulation as fat-laden VLDL particles following the ingestion of fructose, coupled with a fructose-induced reduction in LPL activity, cause sustained postprandial dyslipidemia, thereby augmenting the risk for CV disease ( Fig. 24.10 A,B ).
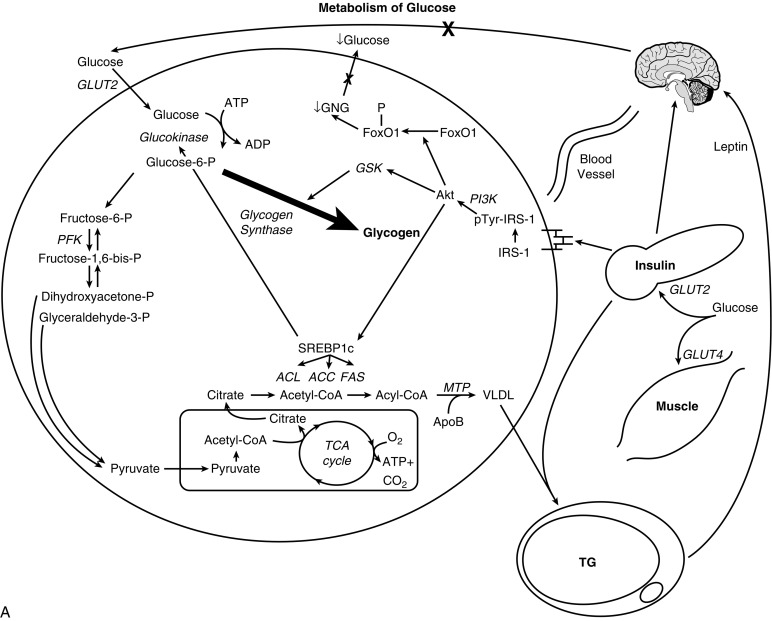
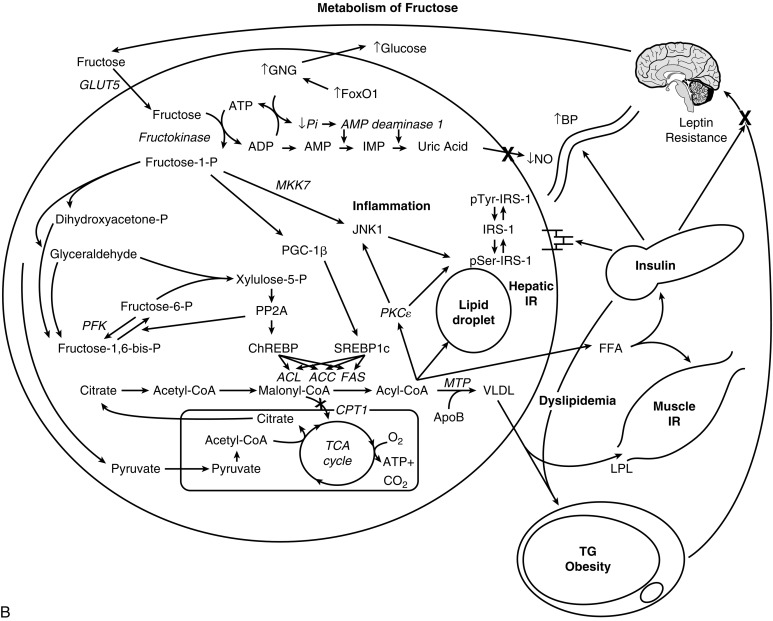
Because of its unique stereochemistry, the ring form of fructose (a five-membered furan with axial hydroxymethyl groups) is under a great deal of ionic strain, which favors the linear form of the molecule, exposing the reactive 2-keto group, which can readily engage in the nonenzymatic fructosylation of exposed amino moieties of proteins via the Maillard reaction, in the same way that the 1-aldehyde position of glucose is reactive. Each Maillard reaction generates one ROS, which must be quenched by an antioxidant or risk cellular damage. In an in vitro study, incubation of hepatocytes with fructose yielded no direct damage; however, when these hepatocytes were preincubated with sublethal doses of hydrogen peroxide to reduce their peroxisomal ROS-quenching ability, fructose then became as hepatotoxic as other organic aldehydes. Mild reduction of fructose in an isocaloric diet intervention for a few days was shown to reduce intrahepatic fat, glucose, and insulin levels indicating its unique metabolic impact, independent of its caloric value.
Branched-Chain Amino Acids
Branched-chain amino acids (BCAAs: valine, leucine, and isoleucine) are essential amino acids that account for more than 20% of the amino acids in the typical “Western diet.” Although normally used for protein biosynthesis and cell growth, when provided in excess they are diverted away from protein synthesis and toward energy utilization.
In the liver, BCAAs increase transcription of ChREBP and SREBP-1c, facilitating DNL. Furthermore, BCAAs limit insulin-induced PI3K signaling and stimulate the activation of the mammalian (aka molecular) target of rapamycin (mTOR), promoting the serine phosphorylation of IRS-1 and impairment of insulin signaling. In addition, just as there are obesity-related changes in adipokines and CV risk markers, there also appear to be obesity-associated changes in BCAA metabolism and subsequent serum levels. In particular, valine and leucine/isoleucine levels have been reported to be 20% and 14% higher, respectively, in subjects with obesity compared with those with normal weight. Mechanistically, this appears to be accounted for by a high rate of flux through the BCAA catabolic pathway, resulting in the increased production of alanine. Because alanine is a highly gluconeogenic amino acid, increased BCAA catabolism may thus contribute to increased hepatic glucose output. Furthermore, the increased α-ketoacids generated by increased flux of the BCAAs through their catabolic pathways also potentially suppress mitochondrial β-oxidation.
Furthermore, chronic BCAA elevation impairs the transport of aromatic amino acids into the brain; the reduced production of serotonin (derived from tryptophan) and catecholamines (derived from phenylalanine and tyrosine) may drive hunger. The “BCAA overload” hypothesis suggests that in the context of a dietary pattern that includes high fat consumption, BCAAs may make an independent contribution to the development of insulin resistance, a hypothesis supported by metabolomics studies demonstrating high BCAA levels in normoglycemic individuals that subsequently develop insulin resistance and diabetes.
Ethanol
Although adult epidemiologic studies associate light to moderate ethanol consumption with improved insulin sensitivity and red wine consumption with reduced CV risk, other cross-sectional and prospective studies implicate a dose-dependent effect of alcohol in metabolic syndrome, and suggest that chronic consumption of large amounts of ethanol worsen insulin sensitivity. Ethanol bypasses glycolysis by being converted by alcohol dehydrogenase-1B to form acetaldehyde, which promotes ROS formation and must also be quenched by hepatic antioxidants, such as glutathione or ascorbic acid. Acetaldehyde is then metabolized by the enzyme aldehyde dehydrogenase-2 to acetic acid, which in turn is metabolized by the enzyme acyl-CoA synthase short-chain family member 2 to form acetyl-CoA. The acetyl-CoA can then enter the mitochondria, or, in the presence of other caloric substrates, it is preferentially used for the synthesis of fatty acids through DNL. The excess malonyl-CoA produced from ethanol metabolism inhibits CPT-1 and thus limits mitochondrial fatty acid β-oxidation. Ethanol also blocks fatty acid β-oxidation by inhibiting PPAR-γ, which suppresses microsomal triglyceride transfer protein, thereby altering the liver’s lipid export machinery. Buildup of intrahepatic lipid metabolites leads to subsequent activation of the enzyme JNK-1 and serine-phosphorylation of the IRS-1, driving further hepatic insulin resistance. Thus ethanol metabolism results in intrahepatic lipid accumulation and liver injury, driving hepatic insulin resistance and promoting the metabolic syndrome. However, although clearly a concern in adults, it is unlikely that ethanol contributes significantly to metabolic syndrome in children.
Calcium and Dairy
There have been several reports of an inverse relationship between dietary calcium and obesity indices. Dietary calcium plays an important role in energy metabolism regulation. Increased calcitriol (1,25-dihydroxyvitamin D) in response to low-calcium diets stimulates Ca 2 + influx in human adipocytes, which may lead to stimulation of lipogenic gene expression and lipogenesis, as well as inhibition of lipolysis. This may result in an expansion of adipocyte triglyceride stores, which can promote adiposity. Increased dietary calcium reduces calcitriol levels and leads to reduction of fat mass without caloric restriction in mice, and this antiobesity effect of dietary calcium is supported by human clinical and epidemiologic studies. Vitamin D deficiency correlates with increasing BMI, especially in African Americans ; however, it is not known if this is caused by substitution of soft drinks for dairy, lactose intolerance, or other factors. One adult study revealed a consistent effect of higher calcium intake on lower body weight and body fat; however, pediatric studies are lacking.
Intestinal Microbiome
Despite establishing a strong association between the gut microbiota and obesity in humans, a causal relationship and discovery of the underlying mechanisms remain to be identified. Studies have shown that fecal transplants from humans with obesity and normal weight to gnotobiotic mice result in adoption of the donor obesity phenotype by the formerly germ-free rodents. This suggests that the microbiome is indeed implicated in the development of obesity, and perhaps in the development of adiposity-related comorbidities. The gut acquires its microbial profile by colonizing bacteria from birth and during the first year of life. Intestinal crosstalk with colonizing bacteria in the developing intestine affects the infant’s adaptation to extrauterine life (immune homeostasis) and may provide protection against disease development (such as obesity) later in life. Disrupted colonization (dysbiosis) caused by maternal dysbiosis, cesarean section delivery, and use of perinatal and neonatal antibiotics may adversely affect the gut development of host defenses and predispose to inflammation rather than to homeostasis, leading to increased susceptibility to development of diseases later in life. It has been shown that the gut microbiome responds to diet, antibiotics, and other external stimuli in ways that impact a variety of metabolic conditions, including obesity and NAFLD. The predominance of certain human intestinal flora species (Firmicutes vs. Bacteroides ) may predispose both animals and humans to obesity, possibly by increasing efficiency of energy absorption ; however, factors that determine their predominance are unknown.
Medications
Numerous medications promote excessive weight gain in children. The most commonly prescribed are pharmacologic doses of glucocorticoids (e.g., prednisone, methylprednisolone, dexamethasone) used for their antiinflammatory and antineoplastic activities. Patients so treated frequently develop obesity, and develop many of the features of Cushing syndrome (e.g., visceral adiposity, hyperlipidemia, hypertension, glucose intolerance), which typify the metabolic syndrome. Sex hormone administration also promotes excessive weight gain, presumably by inducing insulin resistance. In patients with type 1 diabetes, strict glycemic control is usually accompanied by slight overinsulinization of the patient, leading to greater occurrence of mild hypoglycemic episodes requiring nonhunger-driven calorie consumption, and potentially to excessive weight gain. Lastly, more and more children are being placed on the atypical antipsychotics risperidone, olanzapine, quetiapine, clozapine, aripiprazole, and ziprasidone to affect mood and behavior. These antipsychotics are generally associated with weight gain. Studies assessing the weight-protective effects of augmentation therapy with metformin or topiramate demonstrate lower yet still substantial weight gain upon addition of these agents.
Disorders of obesity
The concept that obesity is a phenotype of numerous pathologies is evident from the examination of specific disorders leading to obesity in early childhood ( Box 24.1 , Fig. 24.11 ). Some involve neural mechanisms, others involve classic hormonal mechanisms, whereas others involve dysregulation of increased energy intake, decreased energy expenditure, or increased energy storage at the adipocyte. Remember that even in referral centers for pediatric obesity, children with an “organic” cause of their obesity represent a small minority of the population. Less than 1% will have a “classic” endocrinopathy and less than 3% will have an identifiable genetic etiology in general cohorts with obesity, and approximately 7% will have an endocrinopathy or identifiable genetic cause in pediatric cohorts with severe obesity.
Classic Endocrine Disorders (short stature/growth failure prominent)
- a.
Hypothyroidism
- 1.
Primary
- 2.
Central
- 1.
- b.
Cushing syndrome (glucocorticoid excess)
- 1.
Adrenal adenoma/carcinoma
- 2.
Adrenal micronodular hyperplasia
- 3.
Pituitary ACTH-secreting tumor
- 4.
Ectopic ACTH-secreting tumor
- 5.
Exogenous glucocorticoid administration
- 1.
- c.
Growth hormone deficiency
- d.
Pseudohypoparathyroidism 1a
- 1.
Maternal transmission (AHO + multihormone resistance)
- 2.
Paternal transmission (Pseudopseudohypoparathyroidism, AHO only)
- 1.
Leptin-Melanocortin Pathway Defects (hyperphagia prominent)
- a.
Leptin deficiency
- b.
Leptin receptor deficiency
- c.
POMC mutation (adrenal insufficiency and hypopigmentation/red hair)
- d.
Prohormone convertase-1 deficiency (proinsulin excess)
- e.
Carboxypeptidase E deficiency
- f.
MC3R mutation
- g.
MC4R mutation (tall stature)
- h.
SIM1 mutation (autonomic dysfunction, intellectual disability)
- i.
Melanocortin receptor accessory protein 2 (MRAP2) deficiency
- j.
Brain-derived neurotrophic factor (BDNF) haploinsufficiency/11p14.1 deletion
- k.
NTRK2 mutation
- l.
SH2B1 haploinsufficiency/16p11.2 deletion
Syndromic Obesity Disorders (multiple system involvement and distinct physical features)
- a.
Prader-Willi syndrome
- •
neonatal failure to thrive then rapid weight gain after infancy
- •
hypotonia
- •
intellectual disability
- •
hypogonadism
- •
short stature
- •
- b.
Bardet-Biedl syndrome
- •
retinal dystrophy
- •
polydactyly
- •
intellectual disability
- •
kidney disease
- •
male hypogonadism
- •
- c.
Alström syndrome
- •
retinal dystrophy
- •
hearing loss
- •
pulmonary fibrosis
- •
kidney disease
- •
severe insulin resistance
- •
hypothyroidism
- •
male hypogonadism
- •
- d.
Smith-Magenis syndrome
- •
intellectual disability
- •
maladaptive and self-injurious behaviors
- •
sleep disturbance
- •
- e.
WAGR syndrome
- •
Wilms tumor
- •
aniridia
- •
genitourinary anomalies
- •
range of developmental delays/intellectual disability
- •
reduced pain perception
- •
- f.
Cohen syndrome
- •
microcephaly
- •
hypotonia
- •
intellectual disability
- •
progressive myopia
- •
retinal dystrophy
- •
joint hypermobility
- •
neutropenia
- •
prominent incisors
- •
- g.
Carpenter syndrome
- •
craniosynostosis
- •
intellectual disability
- •
brachydactyly, polydactyly, syndactyly
- •
congenital heart disease
- •
cryptorchidism
- •
umbilical hernia
- •
situs inversus
- •
Hypothalamic Obesity Insulin dynamic disorders
- •
tumor
- •
surgery
- •
radiation
- •
trauma
- •
infiltrative disease
- •
inflammation ROHHAD syndrome (rapid-onset obesity with hypothalamic dysfunction, hypoventilation, autonomic dysregulation, neural crest tumor)
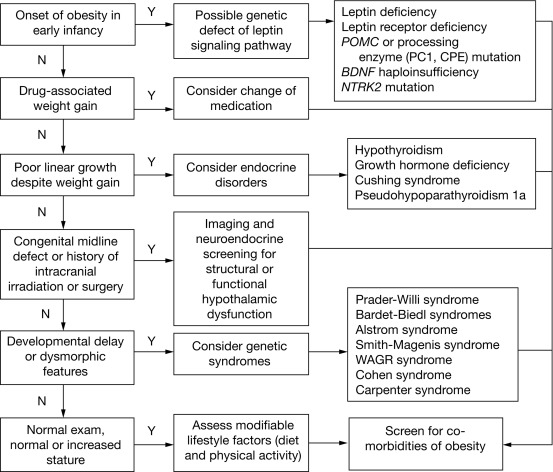
“Classic” Endocrine Disorders With an Obesity Phenotype
In children, linear or statural growth accounts for up to 20% of ingested calories. Endocrine states that allow for normal energy intake for age, but inhibit linear growth, will of necessity lead to excessive energy storage. This is the case for the four “classic” endocrine disorders associated with obesity. These can be distinguished from other causes of pediatric obesity on the basis of their suboptimal growth rate, as opposed to overnutrition, which tends to increase the rate of both growth and skeletal maturation, probably caused, at least in part, by excess insulin cross-reacting with the IGF-1 receptor.
Hypothyroidism. Insufficient triiodothyronine (T 3 ) hormone causes lower REE and decreased physical activity caused by fatigue. Moreover, T 3 is permissive for the anabolic effects of GH. The decrease in total energy expenditure, despite a relatively low caloric intake, promotes persistent energy storage and increases adiposity. Signs, symptoms, and diagnostic evaluation of hypothyroidism are discussed in Chapter 13 . Thyroid hormone replacement is sufficient to increase growth, REE, and physical activity to resolve the obesity over time. Of note, hypothyroidism as a cause of weight gain should not be confused with mild elevations in TSH that occur as a consequence of obesity, thought to be mediated by increased thyrotropin-releasing hormone (TRH) secretion induced by leptin. Thyroid hormone supplementation is generally not indicated in such cases, and TSH usually normalizes with weight loss and reduction in adiposity, which in turn reduces circulating leptin concentrations. Whether thyroid hormone supplementation may aid weight loss or ameliorate CV comorbidities and hepatic steatosis is uncertain.
Glucocorticoid Excess. Cushing syndrome is state of glucocorticoid excess that arrests growth and induces hyperphagia, along with a decrease in REE and physical activity caused by muscle wasting. Exogenous glucocorticoid therapy can result in a similar obesity phenotype. Cushing syndrome is discussed further in Chapter 14 . A reduction of circulating glucocorticoid through medical or surgical treatment reverses obesity, but central adiposity often persists. Although Cushing syndrome as a cause of obesity occurs very rarely, functional hypercortisolism (pseudo-Cushing syndrome) is common in obesity and is thought to be caused by increased HPA-axis activation and greater conversion of inactive cortisone to cortisol by 11βHSD1 in adipose tissue. This chronically higher glucocorticoid tone in obesity and other pseudo-Cushing conditions, such as depression, diabetes, add sleep disorders, may contribute to metabolic health risk and exacerbate visceral adiposity, leading to a viscous cycle of worsening central obesity. Transgenic mice that overexpress 11βHSD1 selectively in adipose tissue develop insulin-resistant diabetes, hyperlipidemia, and hyperphagia. 11βHSD1 enzyme activity is higher in visceral versus subcutaneous adipose tissue and correlated with BMI in normal weight prepubertal children. In adults, however, the associations of 11βHSD1 polymorphisms and BMI or waist : hip ratio were weak at best, and enzyme activity was not elevated in obesity. Nonetheless, specific inhibitors of 11βHSD1 are being investigated as potential drug targets for treatment of obesity and type 2 diabetes.
Growth Hormone Deficiency. Inadequate GH prevents lipolysis and promotes visceral adiposity, although the severity of obesity is usually mild, such that diagnosis of GH deficiency typically precedes the onset of significant obesity. GH deficiency is also associated with fatigue and decreased physical activity. GH deficiency is often accompanied by other pituitary hormone deficiencies (e.g., central hypothyroidism), which can also decrease REE. GH therapy is able to reverse these energy expenditure deficits, increase muscle mass, and promote weight loss. Diagnostic testing for GH deficiency is discussed in Chapter 11 , but it should be mentioned here that obesity itself affects GH secretion such that peak responses during simulation testing are lower in children with obesity compared with those with normal weight. The mechanism for reduced GH secretion in obesity is not known but postulated contributors include hyperinsulinemia and elevated circulating FFAs. Therefore interpretation of GH stimulation testing results should account for altered thresholds in obesity. Moreover, the potential benefit of GH supplementation in relative GH insufficiency remains to be determined.
Pseudohypoparathyroidism Type 1a (PHP1a) and Albright Hereditary Osteodystrophy (AHO). PHP1a and AHO are caused by an autosomal dominant mutation of GNAS1, which encodes the Gsα subunit necessary for peptide hormone signal transduction of GCPRs (affected ligands include parathyroid hormone, TSH, growth hormone-releasing hormone, and α-MSH). Maternal transmission of the GNAS1 mutation leads to PHP1a (multihormone resistance, described further in Chapter 3 and Chapter 20 ) along with AHO, of which a cardinal feature is obesity, caused by reduced MC3R and MC4R anorexigenic/catabolic signaling within the hypothalamus and reduced ability to stimulate cAMP in response to β-adrenergic stimulation within adipocytes. In addition to obesity, other typical manifestations include hypocalcemia, short stature, mild intellectual disability, round face, low nasal bridge, short nose and neck, delayed dental eruption with enamel hypoplasia, short fourth and fifth metacarpals and metatarsals, and short distal phalanges of the thumb. Paternal transmission leads to AHO without multihormone resistance, also known as pseudopseudohypoparathyroidism. Besides hormone replacement and prevention of hypocalcemia, no specific treatments for the obesity aspect of this condition are currently available.
Genetic Obesity Disorders
Approximately 5% to 10% of children with hyperphagia and early-onset (first 3 years of life) obesity are estimated to have an identifiable genetic condition, and the prevalence increases to as high as 22% when screening children with syndromic obesity. Genetic obesity disorders can be categorized as monogenic conditions with obesity as the main phenotype and pleiotropic syndromes that include other characteristics, such as intellectual disability and dysmorphic physical characteristics. There are approximately 80 genetic syndromes that include obesity as a feature, but less than one-fourth of these syndromes have had their genetic etiology fully elucidated. In this chapter, we will focus primarily on the monogenic disorders of the leptin-melanocortin pathway and on a few of the better elucidated pleiotropic syndromes. Further descriptions of the many other syndromes are reviewed in detail elsewhere.
Leptin-Melanocortin Pathway Defects
Since the 1994 discovery of leptin deficiency as the cause of obesity in the ob/ob mouse, the leptin-melanocortin pathway has been extensively elucidated in animal models, whereas in parallel, monogenic defects along this same pathway have been identified in human obesity disorders. Each disorder will be described in sequential order based on the sequence of the pathway steps as shown in Fig. 24.3 . Several pleiotropic obesity syndromes also appear to converge on the leptin-melanocortin pathway, and these conditions will be listed alongside their monogenic disorder counterpart.
Leptin Deficiency. Autosomal recessive mutations of the leptin gene in humans recapitulate the phenotype of the ob/ob leptin-deficient mouse. Only a few such patients have been described, primarily of Pakistani and Turkish descent, and for the most part they were born to consanguineous parents. Birth weight is normal followed by rapid weight gain during infancy and obesity as early as the first few months of life. Hyperphagia manifests at birth as insatiable hunger with constant demands to be fed in infancy and aggressive food-seeking behavior in childhood. The lack of leptin induces the starvation response in the form of central hypothyroidism, lower body temperature, hypogonadotropic hypogonadism, and defective T-cell–mediated immunity. Despite hypothyroidism, normal stature and skeletal maturation are generally preserved until the usual age of puberty because obesity-induced hyperinsulinemia allows excess insulin to cross-react with the IGF-1 receptor to maintain growth. However, because of the important role of leptin in initiating and maintaining puberty, untreated adult patients with leptin deficiency are short because of the lack of a pubertal growth spurt. The diagnosis is made by demonstrating extremely low or unmeasurable serum concentrations of leptin. It should be noted that some mutations of the leptin gene can produce nonfunctional protein that is still detected in standard antibody-based assays so bioactivity measurement may be necessary. In either circumstance of reduced or dysfunctional leptin, treatment with recombinant leptin effectively restores leptin signaling, thereby reducing hyperphagia, resolving obesity, and restoring normal pubertal progression and immune function. Heterozygous carriers of leptin gene mutations display an intermediate phenotype with lower serum leptin concentrations and predisposition for obesity.
Leptin Receptor (LEPR) Deficiency. Autosomal recessive mutations of the leptin receptor gene produce similar symptoms and signs as leptin deficiency. In a large cohort of patients with severe, early-onset obesity, the prevalence of homozygous or compound heterozygous pathogenic variants of LEPR was 3%. Affected individuals had hyperphagia, severe obesity, delayed puberty because of hypogonadotropic hypogonadism and altered immune function. Overall, however, their clinical features were less severe than those of patients with congenital leptin deficiency. Serum leptin concentrations were elevated and correlated with fat mass but shifted higher for the degree of adiposity when compared with control subjects with obesity with normal LEPR . The generally higher circulating leptin concentration in LEPR deficiency is attributed to disruption in the leptin-SNS feedback loop that connects the CNS with peripheral adipocytes. However, because of the fairly high degree of overlap in values, leptin concentrations cannot be used to diagnose LEPR deficiency reliably, so gene sequencing is required. Heterozygous carriers had higher adiposity but were otherwise asymptomatic. Variable presentations depending on type of LEPR mutation have been reported. Three members of a family in France of Algerian ancestry with a homozygous truncating mutation of LEPR (lacking both the transmembrane and the intracellular domains) had additional symptoms of low IGF-1, IGFBP-3, and growth retardation; the cause of these additional features is not known. Treatment of LEPR deficiency requires targeting downstream mediators of the leptin-melanocortin pathway. Because LEPR activation normally would increase POMC and hence the POMC-cleaved product, α-MSH, which binds MC4R, a logical therapeutic target would be melanocortin receptor agonists. One promising candidate drug is setmelanotide, which functions as a biased agonist for MC4R, favoring Gαq signaling regulated by mitogen-activated protein kinase (MAPK), thus avoiding the hypertensive side effect of previously developed MC4R agonists that primary increased Gαs signaling. An initial case report of three patients with homozygous LEPR mutations who received setmelanotide described substantial weight loss with increased skin and hair pigmentation (because of MC1R activation) as the only major side effect. Whether this drug may be beneficial in individuals with heterozygous LEPR mutations remains to be determined.
POMC Deficiency and Splice Site Mutations. Autosomal recessive mutations in the POMC gene that interfere with protein translation or cleavage of the protein into ACTH and α-MSH products results in fair skin and red hair (because of lack of MC1R activation by α-MSH), adrenal hypoplasia and glucocorticoid deficiency (because of lack of MC2R activation by ACTH) with preserved aldosterone and catecholamine production, and obesity (because of lack of MC4R and MC3R activation by α-MSH). Birth weight is normal but hyperphagia drives rapid weight beginning in infancy. Although red hair is a common feature, a Turkish patient with an early nonsense mutation of POMC was reported to have dark hair, indicating that pigmentation can be variable. The diagnosis can be established based on clinical presentation, ACTH deficiency, and hypocortisolemia. Heterozygous carriers have normal pigmentation and cortisol, but higher predisposition for obesity. Methylation variants of POMC in the general population are correlated with individual body weight, suggesting a wider role of POMC in determining obesity risk. A case report in two patients with POMC deficiency showed significant weight loss with setmelanotide treatment, suggesting that MC4R agonism may be beneficial for this condition as well.
Prohormone Convertase-1 Deficiency. Autosomal recessive mutations in the PSCK1 gene that encodes the PC1 enzyme lead to the inability to process various preprohormones to their active ligands, such as POMC to ACTH and α-MSH, proinsulin to insulin, and various gut propeptides to active hormones. Only a handful of patients have been described in the literature, and they are reported as displaying severe early-onset obesity, ACTH deficiency, hypogonadotropic hypogonadism, hyperproinsulinemia, and small intestinal dysfunction because of the inability to cleave intestinal propeptides to their mature form. Hypothyroidism and diabetes insipidus have also been reported. Because of impaired proinsulin cleavage, postprandial hyperglycemia followed by hypoglycemia several hours after a meal is observed. This is caused by initially insufficient insulin secretion followed by reduced clearance of proinsulin, which has mild potency at the insulin receptor, inducing hypoglycemia in the postabsorptive state. The diagnosis can be made by finding extremely high levels of proinsulin, and molecular diagnostics may be required to confirm the gene defect. The potential benefit of using the MC4R agonist setmelanotide to treat obesity in patients with PCSK1 mutations is currently being investigated.
Carboxypeptidase E Mutation. One adult patient with homozygous truncating mutation of the CPE gene has been reported. CPE processes various neuropeptides and prohormones, including POMC and gonadotropins. This patient manifested hyperphagia, severe obesity since childhood, type 2 diabetes, and hypogonadotropic hypogonadism. She also had intellectual disability, which is not a typical characteristic of the other proximal leptin-melanocortin pathway defects discussed thus far. Mice with CPE deficiency display hippocampal degeneration and memory deficits, suggesting a role of CPE in cognitive function.
Melanocortin-3 Receptor Mutation. α-MSH is the endogenous ligand for both MC3R and MC4R, with MC4R being the more critical receptor for regulation of appetite and energy balance whereas MC3R has only a modest role. A heterozygous missense mutation of MC3R was identified in two family members of an ethnically Indian family in Singapore, manifesting as severe early-onset obesity in the child but only mild obesity in the father. The common MC3R variant T6K + V81I is associated with greater adiposity in children and adults. In vitro, this variant appears to decrease MC3R expression. Humanized knock-in mice display increased feeding efficiency and higher triglyceride storage in adipocytes. Diagnosis of MC3R variants can only be made by gene sequencing. No specific treatment currently exists.
Melanocortin-4 Receptor Mutation. Mutations in the MC4R gene appear to account for approximately 5% of severe early-onset obesity cases. Individuals with heterozygous mutations, severe obesity, increased lean mass, increased linear growth, hyperphagia, and severe hyperinsulinemia; individuals with homozygous or compound heterozygous mutations are even more severely affected. Functional severity varies based on the specific mutation, with patients who have mutations retaining residual signaling capacity having a less severe obesity phenotype. Thus mutations in MC4R result in a distinct obesity syndrome that is inherited in a codominant manner. Blood pressure is typically lower compared with control subjects with obesity and normal MC4R , consistent with the role of the melanocortin system in mediating SNS outflow to the renal and CV systems. Intellectual function is typically normal, although a possible association with attention deficit hyperactivity disorder has been reported in children with homozygous mutations. The diagnosis is made by gene sequencing. There is currently no specific treatment for this disorder, but clustered regulary interspaced short palindromic repeat (CRISPR) activation technologies in mice have successfully upregulated the expression of the remaining intact allele in the heterozygous Mc4r knockout to restore normal body weight. Whether this type of gene editing approach can be translatable into humans remains to be determined.
SIM1 Mutation. Single-minded 1 (SIM1) is a human homolog of the Drosophila single-minded gene that is involved in neurogenesis in both species. In humans, SIM1 is involved in the development and function of the PVN, the region of the hypothalamus that expresses MC4R and prodynorphin neurons. SIM1 is believed to function as a downstream mediator of the anorexigenic functions of both these neuronal subpopulations. In a cohort of 2100 individuals with severe obesity, the prevalence of heterozygous SIM1 mutations was 1.3%, and microarray analysis of 279 individuals with syndromic obesity identified one subject with a microdeletion involving SIM1 . Patients with SIM1 haploinsufficiency present with hyperphagia, severe early-onset obesity, normal basal metabolic rate, autonomic dysfunction (lower systolic blood pressure, reduced SNS tone, reduced heart rate variability between sleep and awake states), and varying degrees of neurobehavioral abnormalities and intellectual disability. Increased methylation at the SIM1 locus is associated with higher BMI in adolescents, suggesting that intermediate phenotypes exist in the general population. The diagnosis of SIM1 mutation is made by gene testing. There is currently no specific treatment for this disorder, but CRISPR activation technologies in mice have successfully upregulated the expression of the remaining intact allele in the heterozygous Sim1 knockout to restore normal body weight. Whether this type of gene editing approach can be translatable into humans remains to be determined.
Melanocortin Receptor Accessory Protein 2 ( MRAP2 ) Mutation. MRAP2 protein directly interacts with MC4R and enhances generation of cAMP upon ligand binding of MC4R. Whole-body and brain-specific knockout of Mrap2 in mice causes severe obesity. Heterozygous pathogenic mutations of MRAP2 have been identified in four patients with nonsyndromic severe early-onset obesity without additional syndromic features.
Brain-Derived Neurotrophic Factor Haploinsufficiency. The BDNF gene is located in the 11p14 region. Homozygous loss of function is incompatible with survival but heterozygous loss of BDNF is observed in WAGR syndrome, 11p14 microdeletion, and chromosome 11 paracentric inversion, and BDNF gene variants.
WAGR/11p Deletion Syndrome: The Wilms tumor, aniridia, genitourinary abnormalities, and range of developmental delays syndrome is a rare genetic disorder (prevalence ~ 1 in 1,000,000) caused by contiguous gene deletions on the short arm of chromosome 11. Haploinsufficiency of WT1 and PAX6 , which are involved in genitourinary and eye development, respectively, cause the core features of the syndrome. BDNF resides at 11p14.1, which is 4 Mb from the 11p13 WAGR critical region that contains WT1 and PAX6 , and is included in the deleted segment of approximately half of patients with WAGR syndrome. In a cohort of 33 patients with WAGR syndrome, BDNF haploinsufficiency was associated with higher BMI z-score, fivefold higher frequency of developing childhood obesity (100% vs. 20% for patients with and without BDNF deletion, respectively), reduced serum BDNF concentrations, and higher scores on a parent-reported hyperphagia questionnaire. In this cohort of patients with WAGR syndrome, having BDNF haploinsufficiency versus intact BDNF was also associated with 20-point lower IQ, greater social impairment and higher rate of meeting criteria for autism on the Autism-Diagnostic Interview-Revised, lower parent-reported behavioral responses to typically painful stimuli, and lower self-reported perception of pain in response to hot and cold temperature stimuli.
11p14 Microdeletion: Microdeletions causing BDNF haploinsufficiency while sparring the WAGR region have been reported in 11 individuals with obesity and neurodevelopmental abnormalities. Combining the reports, the 1-Mb common region shared by the deletions of all 11 individuals includes only one gene, BDNF.
11p Paracentric Inversion: Another form of BDNF haploinsufficiency can be caused by loss of expression from one BDNF allele even if the allele is still present. Gray et al. described an 8-year-old girl with a heterozygous paracentric 11p13p15.3 inversion that encompassed BDNF without disrupting the sequence of the gene itself but interfered with expression of the inverted BDNF allele. Her serum BDNF concentration was significantly reduced compared with control subjects with obesity and normal weight, and she displayed hyperphagia, obesity, impaired nociception, and intellectual disability.
BDNF gene mutations/variants: Sequencing of 765 children with early-onset nonsyndromic obesity revealed five heterozygous BDNF rare variants predicted to be deleterious in silico that were not present in 480 control subjects without obesity. The common BDNF Val66Met polymorphism, which impairs activity-dependent BDNF secretion, is linked to binge episodes in bulimia nervosa and binge eating disorder, and BDNF hypermethylation is associated with bulimia nervosa. Associations of BDNF Val66Met with BMI and obesity are inconclusive, but the preponderance suggests that the Met allele is protective. The intronic BDNF rs12291063 variant (homozygous in ~ 10% of individuals with African or Latino ancestry; rare in European non-Hispanic whites) is associated with reduced VMH BDNF expression and increased adiposity. Together these observations indicate that BDNF insufficiency may underlie common, as well as rare, causes of hyperphagia and obesity.
NTRK2 Mutation. The NTRK2 gene encodes the ligand-specific subunit of TrkB, which is the receptor for BDNF. A case report describes one 8-year-old boy with a heterozygous mutation of NTRK2 that prevents autophosphorylation of the receptor upon ligand binding. The child manifested normal weight at birth, hyperphagia and severe weight gain beginning at age 6 months, and neurologic abnormalities including seizures, hypotonia, developmental delay, and decreased nociception.
SH2B1 Haploinsufficiency. Chromosome 16p11.2 has been shown to account for 0.7% of a large cohort of patients with severe obesity and 4% of a pediatric cohort that included children with developmental delay in addition to obesity. A candidate gene for this locus is SH2B1 , which encodes a signaling adapter protein that is needed for BDNF-stimulated neurite outgrowth. Heterozygous mutations in SH2B1 were described in 1.7% of a cohort of patients with severe obesity. The affected patients also displayed neuropsychiatric abnormalities.
Syndromic Obesity Disorders
Several obesity syndromes were included in the earlier section for leptin-melanocortin pathway disorders because the causative genes are thought to be connected with that pathway. Subsequently, additional syndromes with etiologies are not as well delineated.
Prader-Willi syndrome. PWS is a hyperphagic obesity disorder (prevalence ~ 1 in 20,000) caused by lack of expression of paternally derived genes on chromosome 15q11-13, because of heterozygous deletions of paternal alleles (70%), uniparental disomy in which two copies of maternal alleles are inherited (20%–30%), or imprinting defects in which paternal alleles are silenced because of inappropriate methylation (2%–5%). In classical PWS, major diagnostic features include neonatal hypotonia, feeding problems in infancy, rapid weight gain after infancy, hyperphagia, developmental delay, hypogonadotropic hypogonadism, and characteristic facial features (narrow face, almond-shaped eyes, small mouth, thin upper lip, and downturned corners of mouth). Minor diagnostic features include decreased fetal movement, weak cry and lethargy in infancy, behavior problems, sleep disturbance, short stature (GH deficiency), hypopigmentation (in the deletion cases that involve OCA2 , an albinism-related gene in the region), small hands and feet, and skin picking. Other common features include high pain threshold, decreased vomiting, temperature instability, scoliosis, early adrenarche, and unusual skill with jigsaw puzzles.
Attempts to isolate the causative genes for PWS suggest that involvement of more than one gene is likely necessary for the full syndrome to be manifested. Many features of PWS can be observed in patients with inactivating mutations of MAGEL2 , and screening for MAGEL2 mutations is recommended for patients presenting with atypical PWS, also known as Schaaf-Yang syndrome. This syndrome is characterized by infant hypotonia, feeding difficulties, developmental delay, and autism spectrum disorder, but only one-third of patients develop hyperphagia. Magel2 -null mice have neonatal growth retardation, excessive weight gain after weaning, and develop increased adiposity, so these mice can be used as a model to study obesity parameters in PWS. The cause of hyperphagia in PWS is unknown and likely multifactorial. Hyperghrelinemia is observed in patients with PWS, but pharmacologic suppression of ghrelin has not been therapeutically successful at reducing hyperphagia or body weight, calling into question the pathophysiologic role of ghrelin in PWS. Higher circulating EC have also been observed in patients with PWS. Magel2 -null mice have increased expression of EC receptors and administration of an EC antagonist induces weight loss.
Serum leptin concentrations in patients with PWS are appropriately increased in proportion to their higher fat mass and at levels similar to patients with nonsyndromic forms of obesity. Therefore leptin production is preserved in PWS. NPY and AgRP expression appear to be appropriately suppressed in postmortem hypothalamic tissue from patients with PWS, indicating that overexpression of these orexigenic peptides is unlikely to be the cause of hyperphagia in PWS. However, several lines of evidence point to defective signaling along the POMC/CART branch of the leptin-melanocortin pathway. Magel2 -null mice are resistant to the appetite-suppressing effect of leptin administration, with development of leptin insensitivity occurring between 4 to 6 weeks of age, concomitant with a decline in the number of arcuate POMC neurons, suggesting that the transition from failure to thrive in infancy to hyperphagia in early childhood for patients with PWS may be attributable to a neurodegenerative process. Reduced PC1 expression has been reported in induced pluripotent stem cells derived from human patients with PWS, which if also true in vivo, would exacerbate any deficiencies in POMC by further reducing processed α-MSH from PC1 cleavage of POMC. Consistent with the hypothesis that disruption of POMC signaling could be the etiology of hyperphagia in PWS is the observation that patients with PWS have reduced serum and plasma BDNF concentrations compared with control subjects with obesity and normal weight. Considering BDNF’s role as a downstream mediator of MC4R signaling and its function in neurocognition and pain perception, insufficiency of BDNF could potentially account for the hyperphagia, intellectual disability, behavior abnormalities, and high pain tolerance associated with PWS. Although hyperphagia is the primary driver of weight gain, lower lean muscle mass in PWS reduces REE by 40%, further contributing to energy imbalance. GH deficiency also leads to defective lipolysis, further promoting adiposity, but this can be reversed with GH supplementation.
Bardet-Biedl Syndrome (BBS). BBS is an autosomal recessive syndrome (prevalence ~ 1 in 100,000) characterized by hyperphagia, obesity, retinal dystrophy, polydactyly, cognitive impairment, kidney disease, and hypogonadism in males. Over 20 genes encoding proteins involved in the formation, stability, and function of cilia have been implicated in BBS. Mouse models of BBS have defective cilia and impaired leptin receptor trafficking and signaling. These mice are unresponsive to leptin administration with lack of STAT-3 phosphorylation and a lack of reduction in food intake. Hyperleptinemia precedes onset of obesity in some but not all studies, calling into question whether leptin resistance develops before or after the onset of obesity, and the role of cilia in adipocyte differentiation suggests that primary fat deposition may also be a contributing cause of obesity in BBS. In patients with BBS, serum leptin concentrations are higher than those of BMI-matched control subjects, suggesting that in humans, leptin resistance out of proportion to degree of adiposity may play a role in the pathophysiology of ciliopathy-associated obesity. Supporting the hypothesis that insufficient melanocortin signaling contributes to hyperphagia in BBS, a pilot study of the MC4R agonist setmelanotide showed suppression of hunger and induction of weight loss in patients with BBS.
Alström Syndrome. Alström syndrome (AS) is a rare (< 500 reported cases) monogenic form of obesity caused by recessive mutations in the centrosome-body and basal body–associated gene ALMS1 . AS is characterized by retinal dystrophy, sensorineural hearing loss, cardiomyopathy, pulmonary fibrosis, renal disease, childhood obesity, severe insulin resistance, higher susceptibility to type 2 diabetes, elevated triglycerides, and steatohepatitis. Frequent endocrinopathies include hypothyroidism (one-third central, two-thirds primary), central adrenal insufficiency, male hypogonadism (one-third central, two-thirds primary), female hyperandrogenism, and short stature/low IGF-1 concentrations. In contrast with BBS, patients with AS do not have polydactyly and generally retain normal intellectual functioning. The function of the ALMS1 protein is not known but is believed to be important for the formation, stability, and function of cilia. Alms1 knockout mice have reduced number of hypothalamic neuronal cilia, and have hyperphagia and obesity. Because POMC neuronal function is dependent on cilia and intraflagellar transport, reduced POMC function is hypothesized to contribute to the hyperphagia of AS. Consistent with this hypothesis, a pilot study of the MC4R agonist setmelanotide showed suppression of hunger, induction of weight loss, and improved glucose homeostasis in patients with AS.
Smith-Magenis syndrome. SMS (prevalence ~ 1 in 25,000) is caused by heterozygous retinoic acid–induced 1 (RAI1) mutation or deletion. RAI1 is a transcriptional regulator of BDNF involved in craniofacial and nervous system development. In frogs, knockdown of Rai1 using antisense morpholinos results in lower Bdnf mRNA expression and abnormal brain and face development. Heterozygous Rai1 knockout mice have diminished hypothalamic Bdnf expression and display hyperphagia and obesity after age 20 weeks on normal chow, and by 16 weeks on high-fat or high-carbohydrate diets. SMS in human patients is characterized by intellectual disability, maladaptive and self-injurious behaviors, sleep disturbance, and dysmorphic facial features (brachycephaly, broad face, frontal bossing, synophrys, hypertelorism, upslanting eyes, midface hypoplasia with a depressed nasal bridge, a tented upper lip, prognathism, and low-set or abnormally shaped ears). Hyperphagia and obesity are typically not observed until later childhood or adolescence, mimicking the older onset of these symptoms in mice, and are more pronounced in patients with Rai1 mutations compared with those with deletions. Examination of functional effects of mutations found in patients has revealed that the mutated RA1 protein fails to localize to the nucleus and does not activate expression of a reporter gene driven by an endogenous BDNF promoter. Presence of the mutated protein may have a dominant negative effect on the remaining normal protein, thus leading to a more severe obesity phenotype in patients with mutations.
Cohen Syndrome. Cohen syndrome (< 1000 cases reported) is caused by autosomal recessive mutations in VPS13B and characterized by microcephaly, hypotonia, intellectual disability, progressive myopia, retinal dystrophy, joint hypermobility, neutropenia, truncal obesity that develops in later childhood, insulin resistance/type 2 diabetes, and dysmorphic features (thick hair and eyebrows, long eyelashes, downslanting and wave-shaped palpebral fissures, bulbous nasal tip, smooth or shortened philtrum, prominent upper central teeth, open mouth, narrow hands and feet, and slender fingers). VPS13B encodes a Golgi apparatus protein that is involved in protein glycosylation and intracellular transport in neurons and adipocytes.
Carpenter Syndrome. Carpenter syndrome (< 100 cases reported) is caused by autosomal recessive mutations in RAB23 (involved in vesicle trafficking) or MEGF8 (involved in cell adhesion) and is characterized by acrocephalic or cloverleaf craniosynostosis, intellectual disability, childhood-onset obesity, flat nasal bridge, downslanting palpebral fissures, low-set and abnormally shaped ears, micrognathia, small primary teeth, cutaneous syndactyly of third and fourth fingers, brachydactyly, polydactyly, umbilical hernia, hearing loss, heart defects, deformed hips, kyphoscoliosis, genu valgum, cryptorchidism, situs inversus, dextrocardia, and transposition of the great arteries.
Hypothalamic Obesity
Hypothalamic damage can occur because of CNS tumor, surgery, radiation, trauma, inflammation, or infiltrative diseases. Injury to the VMH and ARC of the MBH are the critical regions that lead to the phenomenon of hypothalamic obesity, which is characterized by extremely rapid weight gain. For example, it is typical in the first 6 to 12 months after craniopharyngioma surgery for tumors involving the floor of the third ventricle (the hypothalamic homeostatic centers described earlier) for patients to increase weight by greater than 50%. Mechanistically, it has been well known that bilateral electrolytic lesions or deafferentation of the VMH in rats leads to intractable weight gain, even upon food restriction. Originally, weight gain was thought to be solely a problem with hyperphagia and increased energy storage. However, we now understand that a dysfunction of leptin-melanocortin pathway signaling can alter both the afferent and efferent pathway of energy balance and lead to severe and intractable weight gain that is caused by more than just overeating. The hypothalamic insult prevents the integration of peripheral afferent signals, and therefore the CNS is unable to detect nutrient sufficiency within the periphery and therefore responds as if in the starvation state. Caloric restriction alone is insufficient because patients also have reduced sympathetic activity, which reduces energy expenditure and lipolysis, and heightened parasympathetic (vagal) tone, which promotes insulin hypersecretion and energy storage. In both animals and humans, vagal hyperreactivity can be prevented by pancreatic vagotomy. Therapy for this disorder remains extremely problematic, as the brain seems to be “locked” in an orexigenic balance favoring energy consumption and storage. As the crucial period of weight gain is within the first year following the brain insult, it is crucial to focus on preventive measures during this narrow window of opportunity. Comprehensive treatment should include psychologic care to address cognitive, behavior, and sleep disturbances that commonly occur; endocrine hormone replacement for deficiencies; and intensive lifestyle (nutrition and exercise) management. Medical therapies, such as insulin sensitizers, insulin secretion blockers, GLP-1 analogues, stimulants, and oxytocin have all been investigated and each show modest benefits, but the intractable nature of the disease makes management exceedingly challenging. It is accepted practice today that all efforts should be made to avoid surgical trauma of the hypothalamic homeostatic centers, even at the price of leaving remnant tumor tissue (in cases of craniopharyngioma which typically involves this region).
ROHHAD (rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysfunction) syndrome is a form of hypothalamic obesity that is not well understood and has a very poor prognosis. Median age of diagnosis is 4 years and presents as extremely rapid weight gain in an otherwise previously healthy child. ROHHAD appears to be a progressive neurodegenerative condition that evolves from hyperphagic obesity to global hypothalamic dysfunction (including GH deficiency, hypogonadotropic hypogonadism, central hypothyroidism, and ACTH abnormalities), central hypoventilation or altered respiratory control, autonomic dysfunction (bradycardia requiring pacemaker placement), thermal dysregulation, and risk for the development of tumors of neural crest origin (e.g., ganglioneuromas and ganglioneuroblastomas). Although BDNF and NTRK2 have been screened as candidate genes for ROHHAD, no mutations in these genes have been identified to date. However, one patient, an 11-year-old boy with BMI of 62 kg/m 2 with a clinical diagnosis of ROHHAD, was found to have a heterozygous truncating mutation of RAI1, which is a transcriptional regulator of BDNF and is implicated as the causative gene for SMS. In addition to severe obesity, this patient also exhibited intellectual disability, autism spectrum disorder, dysmorphic facial features (macrocephaly, hypertelorism, flat nasal bridge, prominent forehead, and anteverted nares), high pain tolerance, excessive sweating, lack of fever with infections, obstructive and central sleep apnea, and hypoventilation leading to tracheostomy placement. Pituitary hormone abnormalities and neural crest tumors were lacking in this patient. Thus he could be categorized as atypical ROHHAD or possibly atypical SMS, and screening for RAI1 haploinsufficiency should be considered in the differential for patients presenting with features of ROHHAD. Without treatment, ROHHAD has a high mortality rate from respiratory failure. Immunomodulator therapies have been attempted and show some benefit in slowing disease progression but there are no curative treatments known.
Evaluation and treatment of pediatric obesity
Diagnostic Approach
The key to successful obesity therapy is accurate diagnosis. Our diagnostic armamentarium is not yet fully developed, so matching treatment to diagnosis is still uncertain. Specific points in the evaluation and their rationale are listed in Table 24.1 . In eliciting the history, birth weight, parent’s BMIs, exposure to gestational diabetes, prematurity, history of breastfeeding, and neonatal complications (especially CNS injury) are all relevant. The younger the patient’s obesity is noted, the more likely an organic reason will be identified. Neurodevelopmental abnormalities and signs of dysmorphism may signify the need for a genetic referral. The medication list must be reviewed, especially for glucocorticoids and atypical antipsychotics. Orthopedic pain, headache, and snoring must be assessed. Dietary history must include skipping breakfast, daily ingestion of sodas and juices, and frequency and type of snacking. A corollary is the number of caretakers of the child because this increases stress, family chaos, and lack of child supervision.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


