Abstract
Obesity, defined as an excess fat mass, results from individual genetic and biological susceptibilities in response to the current weight-gain promoting environment. There is thus a synergistic relationship between genes and environment. Depending on the genes involved, different clinical situations are described: (1) monogenic obesity defined as rare and severe early-onset obesity associated with endocrine disorders, which are mainly due to autosomic recessive mutations in genes of the leptin-melanocortin axis – in some cases, such as leptin deficiency, specific treatments are efficient; (2) melanocortin 4 receptor-linked obesity characterized by the variable severity of obesity and the absence of additional phenotypes, which are involved in 2–3% of cases and eventually may benefit from new developed drugs (MC4R agonists); and (3) syndromic obesity where patients are clinically obese, and additionally distinguished by mental retardation, dysmorphic features, and organ-specific developmental abnormalities. No specific treatments are recommended except for hormonal substitution if necessary (growth hormone or gonadotrope deficiency).
Keywords
obesity, genetics, melanocortins, leptin, melanocortin 4 receptor, proopiomelanocortin
Introduction
Background, Incidence, Prevalence
According to the World Health Organization ( http://www.who.int.easyaccess2.lib.cuhk.edu.hk ), there are an estimated 1.5 billion adults who are overweight (body mass index (BMI) ≥25 kg/m 2 ), and 500 million of these are considered clinically obese (BMI ≥30 kg/m 2 ). The worldwide prevalence of childhood overweight and obesity also increased from 4.2% in 1990 to 6.7% in 2010, but has stabilized in recent years. Obesity, defined by an excess fat mass having an impact on health, is characterized by a wide phenotype heterogeneity. There are also individual differences in progression of weight (i.e., different trajectories) and risk of associated comorbidities. It is now well accepted that the development of obesity stems from the interaction of multiple environmental factors (such as overeating and/or reduction in physical activity) with genetic factors. Numerous epidemiological and intervention studies carried out in different cohorts (twins brought up together or separately, adopted children, nuclear families, etc.) have recognized the role of individual genetic and biological susceptibilities in response to the current weight-gain promoting environment. The severity of obesity will be thus determined by the impact of the environment on the genetic background of each individual.
While obesity was first thought to be a disease that obeys the rules of Mendelian inheritance, new technologies paint a far more complicated picture of this complex disorder. Obesity due to a single, naturally occurring dysfunctional gene (i.e., monogenic obesity) is both severe and rare (from <1% to 2–3% depending on the gene) when compared to the more common forms in which numerous genes make minor contributions in determining phenotypic traits in large populations (i.e., polygenic obesities).
Clinical Presentation
Several clinical presentations are described in obesity depending on the involved genes:
- 1.
Monogenic obesity , described as rare and severe early-onset obesity associated with endocrine disorders. This is mainly due to mutations in genes of the leptin-melanocortin axis involved in food intake regulation (genes of leptin ( LEP ) and its receptor ( LEPR ), proopiomelanocortin ( POMC ), proconvertase 1( PC1 ), etc.). New mutations were identified in recent years.
- 2.
Melanocortin-4 receptor (MC4R)-linked obesity , characterized by a variable severity of obesity and the absence of very noticible phenotype. This is responsible for 2–3% of obesity in adults and children.
- 3.
Syndromic obesity , corresponding to obesity associated with other genetic syndromes, as reviewed elsewhere. Patients are clinically obese and additionally distinguished by mental retardation, dysmorphic features, and organ-specific developmental abnormalities.
- 4.
Polygenic obesity , which is the more common clinical situation (>95% of cases). Here each susceptibility gene, taken individually, would only have a slight effect on weight. The cumulative contribution of these genes would become significant only in an “obesogenic lifestyle” (such as overfeeding, sedentariness, stress, and others). This part will not be developed in this chapter.
Genetic pathophysiology
Monogenic Obesity
At least 200 cases of human obesity have been associated with a single gene mutation. These gene mutations all lie in an increased number of genes, but the monogenic forms of obesity remain rare in the obese population. Unlike syndromic obesity, the reason why excess body fat mass develops in these subjects is quite well understood since the genetic anomalies affect key factors related to the leptin and the melanocortin pathways well-known to be pivotal in energy balance regulation ( Fig. 21.1 ). Most of the severe forms of childhood obesity are due to single gene mutations; new ones are regularly discovered in different families.
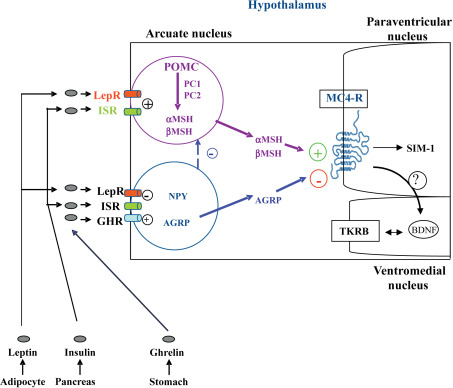
The hypothalamic leptin/melanocortin pathway is activated following the systemic release of the adipokine LEP and its subsequent interaction with its receptor LEPR, located on the surface of neurons of the arcuate nucleus. The downstream signals that regulate satiety and energy homeostasis are then propagated via POMC, cocaine-and-amphetamine-related transcript (CART), and the melanocortin system. While POMC/CART neurons synthesize the anorectic peptide α-melanocyte stimulating hormone (α-MSH), a separate group of neurons express the orexigenic neuropeptide Y (NPY) and the agouti-related protein (AGRP), which acts as a potent inhibitor of melanocortin 3 (MC3R) and MC4R receptors. The nature of the POMC-derived peptides depends on the type of endoproteolytic enzyme present in the specific brain region. In the anterior pituitary, the presence of the PC1 enzyme produces adrenocorticotropic hormone (ACTH) and β-lipotropin peptides, while the combined presence of PC1 and PC2 in the hypothalamus controls the production of α-, β-, γ-MSH, and β-endorphins.
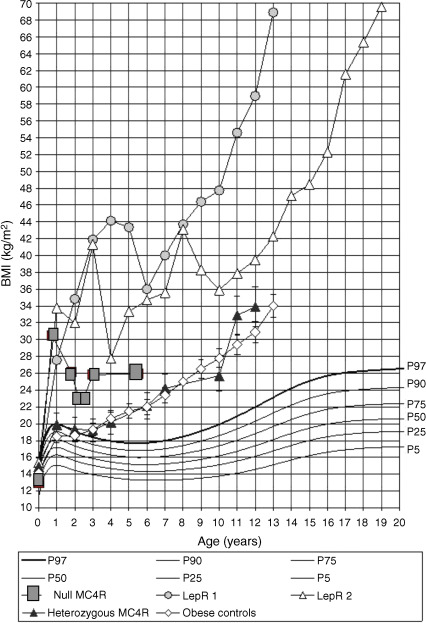
Mutations in human genes coding for LEP, LEPR, POMC, and PC1 lead to severe obesity occurring soon after birth, with generally complete penetrance and autosomal-recessive transmission ( Table 21.1 ). Patients carrying mutations show a rapid and dramatic increase in weight, as illustrated by the weight curve of LEPR deficient subjects ( Fig. 21.2 ). Evaluating body composition in some LEPR mutation carriers shows a large amount of total body fat mass (>50%) but resting energy expenditure remains related to the level of body corpulence. Feeding behavior is characterized by major hyperphagia and ravenous hunger. Surprisingly, a LEP-deficient Austrian girl has been recently described with more moderate obesity (BMI 31.5 kg/m 2 ), despite an increased consumption of calories in a test meal. The phenotype was explained by extremely low daily calorie intake. Even if one takes into account a substantial underreporting, this observation might suggest that despite leptin deficiency, it was possible to control energy intake and thus to prevent extreme obesity. In that specific case the parents’ role was described as determinant. They provided a favorable environment by vigorously controlling the patient’s eating behavior from early infancy onward. A further explanation might be related to the different genetic backgrounds of different subjects with LEP or LEPR deficiency. However, despite this particular case, severe early-onset obesity with major hyperphagia is recognized as a main clinical presentation of LEP or LEPR deficiency.
| Gene (references) | Mutation type | Obesity | Associated phenotypes |
|---|---|---|---|
| Leptin | Homozygous mutation | Severe, from the first days of life | Gonadotropic and thyrotropic insufficiency |
| Leptin receptor | Homozygous mutation | Severe, from the first days of life | Gonadotropic, thyrotropic, and somatotropic insufficiency |
| Proopiomelanocortin (POMC) | Homozygous or compound heterozygous | Severe, from the first months of life | ACTH insufficiency. Mild hypothyroidism and ginger hairs if the mutation leads to the absence of POMC production |
| POMC but in the β-MSH coding region | Heterozygous non synonymous mutations | Severe obesity occurring in childhood | Rapid size growth |
| Proprotein convertase subtilisin/kexin type 1 (PCSK1 or PC1) | Homozygous or compound heterozygous | Severe obesity occurring in childhood | Adrenal, gonadotropic, somatotropic, and thyrotropic insufficiency. Postprandial hypoglycemic malaises. Central diabetes insipidus |
| Single-minded 1 (SIM1) | Translocation between chr 1p22.1 and 6q16.2 in the SIM 1 gene | Severe obesity occurring in childhood | Inconstantly neurobehavioral abnormalities (including emotional lability or autism-like behavior) |
| Neurotrophic tyrosine kinase receptor type 2 (NTRK2) | De novo heterozygous mutation | Severe obesity from the first months of life | Developmental delay. Behavioral disturbance. Blunted response to pain |
| Dedicator of cytokinesis 5 (DOCK5) | Variable number tandem repeats (VNTRs) | Childhood and adult severe obesity | – |
| Kinase suppressor of Ras2 (KSR2) | Heterozygous frameshift, nonsense, or missense variants | Severe obesity | Hyperphagia in childhood. Low heart rate. Reduced basal metabolic rate. Severe insulin resistance |
| Tubby-like protein (TUB) | Homozygous frameshift mutation | Early-onset obesity | Night blindness, decreased visual acuity, and electrophysiological features of a rod cone dystrophy |
Associated to severe early-onset obesity with major hyperphagia, hypogonadotrophic hypogonadism, and thyrotropic insufficiency complete the patient phenotype. Insufficient somatotrophic secretion, leading to moderate growth delay, is also described in some patients with a LEPR mutation. In LEP deficient subjects, it was described as a high rate of infection, particularly recurrent respiratory tract infections, associated with a deficiency in T-cell number and function. In individuals with leptin deficiency, either due to LEP or LEPR mutations, no pubertal development was observed, while in others, there is evidence of spontaneous pubertal development, suggesting a recovery of hormonal functions with time. For example, the follow-up of the initially described LEPR deficient sisters revealed the normalization of thyroid mild dysfunction at adult age and a normal spontaneous pregnancy.
Measurement of circulating leptin may help the diagnosis in some cases; it is undetectable in LEP mutation carriers. However, leptin levels can be correlated to fat mass or extremely elevated in LEPR mutation carriers. Thus, LEPR gene screening might be considered in subjects with the association of severe obesity with endocrine dysfunctions such as hypogonadism but with leptin related to corpulence level.
Obese children with a complete POMC deficiency have ACTH deficiency, which can lead to acute adrenal insufficiency from birth. These children display a mild central hypothyroidism that necessitates hormonal replacement and sometimes alterations in the somatotropic and gonadotropic axes. The reason for these endocrine anomalies is unknown, even if the role of melanocortin peptides in influencing the hypothalamic pituitary axis has been proposed. Children have inconstantly ginger hair due to the absence of α-MSH, which activates the peripheral melanocortin receptor type 1 (MC1R) (involved in pigmentation). Several observations suggest that the skin and hair phenotype might vary according to the ethnic origin of POMC mutation carriers. The modifications in color hair, adrenal function, and body weight are consistent with the lack of POMC-derived ligands for the melanocortin receptors MC1R, MC2R, and MC4R, respectively.
Patient carriers of a rare mutation in the proprotein convertase subtilisin/kexin type 1 ( PCSK1 ) gene leading to PC1 deficiency have, in addition to severe obesity, postprandial hypoglycemic malaises, fertility disorders due to hypogonadotrophic hypogonadism, central hypothyroidism, and adrenal insufficiency secondary to lack of POMC maturation. The delayed postprandial malaises are explained by the accumulation of proinsulin through lack of PC1, which is involved in the synthesis of mature insulin from proinsulin. The absence of POMC maturation due to PCSK1 mutation causes a dysfunction in the melanocortin pathway that explains the obese phenotype. In patients suffering from a congenital PC1 deficiency, severe and rebel diarrhea due to a defect in intestinal absorption is described, secondary to lack in mature GLP-1 (glucagon-like peptide-1). The processing of prohormones, progastrin and proglucagon, is altered, explaining, at least in part, the intestinal phenotype and also suggesting a role for PC1 in absorptive functions in the intestine. Recently, in a proband who was compound heterozygous for a maternally-inherited frameshift mutation and a paternally-inherited 474-kb (kilobase) deletion that encompasses PCSK1 and in 13 children with total PC1 deficiency, persistent polydipsia and polyuria were noted, as the disease progressed, due to the development of central diabetes insipidus improved by oral desmopressin. These observations suggest that PCSK1 may be involved in the full functioning or central sensing of osmolality in humans. Growth hormone deficiency was also noted in the 13 children with complete PC1 deficiency.
These studies have played an important part in confirming the critical role of the leptin and melanocortin pathways in controlling food intake and energy expenditure, as well as their strong implication in controlling endocrine pathways. Furthermore, these studies encouraged the pursuit of screens for genes encoding proteins acting both upstream and downstream of the G-protein coupled receptor MC4R ( Table 21.1 and Fig. 21.1 ). Several additional genes, implicated in the development of the hypothalamus and the central nervous system, have been found to cause monogenic obesity in humans. A deletion of the SIM1 ( single-minded homolog 1 ) gene, located on the 6q chromosome, secondary to a de novo translocation between 1p22.1 and 6q16.2 chromosomes, was identified in a girl with early-onset obesity (since first months of life) associated with hyperphagia and food impulsivity. She had a rate of early weight gain comparable to the weight curve of LEP and LEPR -deficient children. Izumi et al. identify an interstitial 6q deletion, including the SIM1 gene in a subject with Prader–Willi-like features (neonatal hypotonia, dysmorphy, developmental delay, early-onset obesity, short stature, hypopituitarism). SIM1 encodes a transcriptional factor implicated in the development of the hypothalamic paraventricular nucleus. It plays a role in the melanocortin signaling pathway and appears to regulate feeding rather than energy expenditure. The sequencing of the coding region of SIM1 , in 2,100 unrelated patients with severe, early-onset obesity and in 1,680 unrelated population-based controls, identified 13 different heterozygous variants in 28 severely obese patients. Variants carriers exhibited severe obesity, increased ad libitum food intake at a test meal, normal basal metabolic rate, and inconstantly neurobehavorial phenotype (impaired concentration, memory deficit, emotional lability, or autistic spectrum behavior). Nine of the 13 variants significantly reduced the ability of SIM1 to activate a SIM1 -responsive reporter gene. These mutations cosegregated with obesity in extended family studies with variable penetrance. Rare variants in SIM1 should be considered in patients with hyperphagic early-onset obesity, associated or not to Prader–Willi-like syndrome features (including severe obesity that starts between 1 and 2 years of age) and/or in patients with associated neurobehavioral abnormalities (including emotional lability or autism-like behavior).
A decreased expression of the brain-derived neurotropic factor (BDNF) was found to regulate eating behavior. BDNF, encoded by the NTRK2 gene ( neurotrophic tyrosine kinase receptor type 2 ) and its associated tyrosine kinase receptor (TRKB) are both expressed in the ventromedial hypothalamus. They have been attributed a role downstream of MC4R signaling implicated in feeding regulation. A de novo heterozygous mutation in the NTRK2 gene was described in an eight-year-old boy with severe early-onset obesity, mental retardation, developmental delay, and anomalies of higher neurological functions like the impairment of early memory, learning, and nociception. Other mutations in NTRK2 were found in patients with early-onset obesity and developmental delay, but their functional consequences and their implication in obesity are yet to demonstrate. In vitro studies of some but not all mutations have suggested that these mutations could impair hypothalamic-signaling processes.
Finally, the contribution of copy number variants (CNVs) to complex disease susceptibility, such as severe obesity, has been the subject of debates in recent years. Variable number tandem repeats (VNTRs) constitute a relatively underexamined class of genomic variants in the context of complex diseases. Rare CNVs have been shown to be responsible for severe highly penetrant forms of obesity, initially observed in 31 subjects who were heterozygous for deletions of at least 593 kb at 16p11.2 locus. In addition to obesity, these carriers presented with congenital malformations and/or developmental delay. The phenotype may result from the haploinsufficiency of multiple genes that impact on pathways central to the development of obesity. An investigation of a complex region on chromosome 8p21.2 encompassing the DOCK5 ( dedicator of cytokinesis 5 ) gene has shown a significant association of the DOCK5 VNTRs with childhood and adult severe obesity. More systematic investigation of the role of VNTRs in obesity had to be performed to study their relatively unexplored contribution to this disease and their potential link with the leptin/melanocortin pathway.
Melanocortin 4 and 3 Receptor-Linked Obesities
The obesities linked to MC4R and MC3R mutations can be placed between the rare forms of monogenic obesity with complete penetrance and the polygenic forms of common obesity.
Considering the pivotal role of the melanocortin pathway in the control of food intake, the MC4R gene is a major candidate gene in human obesity ( Fig. 21.1 ). Since 1998, its genetic evaluation revealed that MC4R -linked obesity is the most prevalent form of genetic obesity identified to date. It represents approximately 2–3% of childhood and adult obesity with currently 166 different mutations described in different populations (European, North American, and Asian). They include frameshift, inframe deletion nonsense, and missense mutations located throughout the MC4R gene ( Fig. 21.3 ). The frequency of heterozygosity for these mutations in (extremely) obese individuals cumulates to approximately 2–5%. In addition, the frequency of such heterozygous carriers in nonobese controls or in the general population is about 10-fold lower than in the cohorts of obese patients.
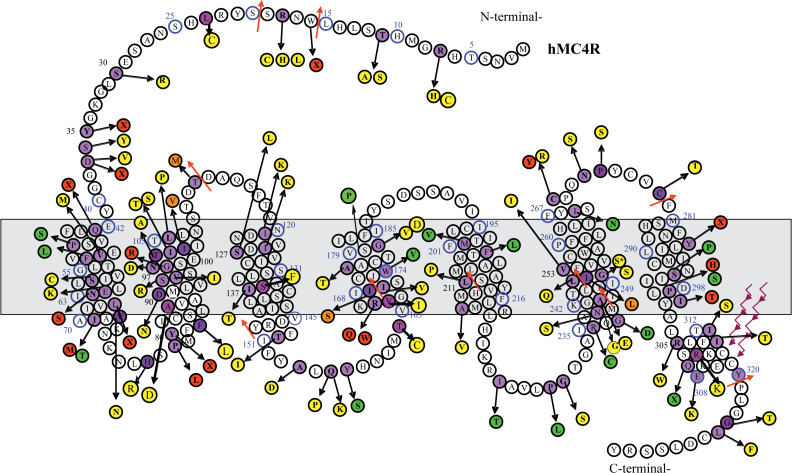
In contrast with rare monogenic obesities, even a meticulous clinical analysis does not easily detect obesity stemming from MC4R mutations because of the lack of additional obvious phenotypes. In families with MC4R -linked obesity, obesity tends to have an autosomal dominant mode of transmission, but the penetrance of the disease can be incomplete and the clinical expression variable (moderate to severe obesity), underlying the role of the environment and other potentially modulating genetic factors. Homozygous or compound heterozygous carriers of MC4R mutations are very rare. Four carriers of homozygous null mutations in the MC4R gene have been detected, and as expected from a dominant condition, obesity is developed earlier in life and is more severe than for heterozygous carriers, but it does not display any additional unrelated phenotypes. In heterozygous MC4R mutations carriers, the onset and severity of obesity vary and are related to the severity of the functional alteration caused by the mutation.
Many authors agree that MC4R mutations facilitate early-onset obesity. MC4R mutations carriers display increased linear growth, in particular in the first five years of life but appear to be taller as adults only in rare cases. This trend is often observed in overweight and obese children. Assessment of body composition in these patients demonstrates increase in both fat and lean mass. One study performed in English children with MC4R mutations has suggested that bone mineral density and size increase. This potential increase of bone density may be explained, at least in part, by a decrease in bone resorption, as illustrated by decreases in bone resorption markers in the serum of patients with MC4R homozygous and heterozygous mutations. Obese children carrying MC4R mutations have a marked hyperphagia that decreases with age when compared to their siblings, while in both children and adults, no evidence has been found for a decreased metabolic rate in these patients. Meanwhile, the association between “binge eating” disorder and MC4R gene sequence changes has not been confirmed. Adult MC4R mutation carriers do not have an increased prevalence of diabetes or other obesity complications. In UK children, fasting insulinemia was found to be significantly elevated in MC4R mutation carriers, particularly before the age of 10 years when compared to age, sex, and BMI matched control. This hyperinsulinemia has not been consistently observed in children and in adults. Finally, with respect to endocrine function, hypothalamopituitary axis and reproductive axis, as well as thyroid function, are normal in MC4R mutation carriers.
The role of MC4R mutations in cases of human obesity relies on two main arguments based on the frequency of MC4R mutations in different populations and their in vitro functional consequences. Firstly, MC4R mutations are more abundant in obese populations. Functional mutations have also been reported in nonobese subjects but to a significantly lesser frequency (<1%). Secondly, investigating the molecular mechanisms by which loss-of-function mutations in MC4R cause obesity have suggested a panel of functional anomalies: abnormal MC4R membrane expression, defect to the agonist response, and a disruption in the intracellular transport of this protein. Normally, after ligand binding, MC4R activation stimulates Gs protein, leading to a subsequent increase in cAMP levels; however, the production of intracellular cAMP in response to α-MSH peptides demonstrated a broad heterogeneity in the activation of the different MC4R mutants in response to α-MSH, ranging from normal or partial activation to a total absence in activation. The intracellular transport defect of the mutated receptor, by intracytoplasmic retention, has been described for the majority of MC4R mutations found in childhood obesity, but also in adults. This mechanism explains the impaired response to agonists. In addition, MC4R has a constitutive activity, meaning a basal activity not necessitating the presence of a ligand, for which agouti-related peptide (AGRP) acts as an inverse agonist. In the absence of the ligand, MC4R has an inhibitory action on food intake. The systematic study of basal activity of some mutations has shown that an alteration in this activity may be the only functional anomaly found, in particular for mutations located in the N-terminal extra-cytoplasmic part of the receptor. A tonic satiety signal, provided by the constitutive activity of MC4R, could be required in the long-term regulation of energy balance. It is accepted that MC4R mutations cause obesity by a haploinsufficiency mechanism rather than a dominant negative activity. While the roles of homo- and heterodimerization in G protein synthesis and maturation are emphasized, some dominant negative effects of MC4R mutations might not be excluded.
The MC3R has also been involved in obesity. MC3R deficient ( MC3R –/– ) mice have increased fat mass, reduced lean mass, and higher feed efficiency than wild-type littermates, despite being hypophagic and maintaining normal metabolic rates. In humans, strong evidence of a causative role for MC3R mutations is still lacking. Some mutations, leading to amino acid changes in the receptor, have been described in a group of 290 obese subjects, but the total prevalence of rare MC3R variants was not significantly different between cohorts of severely obese subjects and lean controls. No specific phenotype of MC3R mutations has been identified.
Obesity Syndromes
There are between 20 and 30 Mendelian disorders in which patients are clinically obese with such additional phenotypes as mental retardation, dysmorphic features, and organ-specific developmental abnormalities. These syndromes arise from discrete genetic defects or chromosomal abnormalities and are both autosomal and X-linked disorders. The most common and well-known disorders are Prader–Willi and Bardet–Biedl syndromes, but many others have been reported ( Table 21.2 ). The Online Mendelian Inheritance in Man database provides access to their clinical descriptions (OMIM; http://www-ncbi-nlm-nih-gov.easyaccess2.lib.cuhk.edu.hk/omim/ ).
| Syndrome | Clinical features in addition to obesity | Locus | Gene |
|---|---|---|---|
| AUTOSOMAL DOMINANT | |||
| Prader–Willi syndrome (PWS) | Neonatal hypotonia, mental retardation, facial dysmorphy, hypogonadotrophic hypogonadism, short stature | Lack of the paternal segment 15q11.2-q12 (microdeletion or maternal disomy) | Unknown SRNPN region |
| Albright hereditary osteodystrophy | Short stature, skeletal defects, facial dysmorphy, endocrine anomalies | 20q13.2 | GNAS1 |
| AUTOSOMAL RECESSIVE | |||
| Bardet–Biedl syndrome (BBS) | Mental retardation, dysmorphic extremities, retinal dystrophy or pigmentary retinopathy, hypogonadism, kidney anomalies (structural abnormalities or functional renal impairment) | 1q13 (BBS1), 16q21 (BBS2), 3p13 (BBS3), 15q22 (BBS4), 2q31 (BBS5), 20p12 (BBS6), 4q27 (BBS7), 14q32 (BBS8)…. | BBS 1–19 |
| Alström syndrome | Retinal dystrophy, neurosensory deafness, diabetes | 2p13 | ALMS1 |
| Cohen syndrome | Prominent central incisors, dysmorphic extremities, ophthalmopathy, microcephaly, cyclic neutropenia | 8q22 | COH |
| X-LINKED | |||
| Borjeson–Forsmman–Lehman syndrome | Mental retardation, hypogonadism, facial dysomoprhy with large ears | Xq26 | PHF6 |
| Fragile X syndrome | Mental retardation, hyperkinetic behavior, macroorchidism, large ears, prominent jaw | Xq27.3 | FMR1 |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


