Success in therapy for adults with acute lymphoblastic leukemia (ALL) has lagged behind that of children with ALL. However, the application of pediatric-intensive regimens to young and middle-aged adults suggests improved outcomes without excessive toxicity. A promising area in the treatment of adult ALL is the addition of monoclonal antibodies in various forms to chemotherapy in adults with ALL. New chemotherapeutic approaches show promise, and early-phase clinical trials with these drugs are under way.
The notable success of therapy for acute lymphoblastic leukemia (ALL) in children, for whom cure rates now exceed 80% to 90%, is even more remarkable when one considers that much of this success has occurred through alteration in drug dosing and schedule rather than the introduction of new agents. In adults, morphologic complete remission rates are now more than 90%. Relapse is common and cure rates, up until recently, have only been in the 30% to 40% range. Despite these poor results, treatment outcomes in all age groups with ALL have improved when one compares results from the early 1980s to the early 2000s. The poorer outcomes in adults are multifactorial, but largely relate to a higher incidence of poor-risk cytogenetic features and lessened ability to tolerate intensive chemotherapy regimens that are often used in pediatric patients.
Patients presenting to adult oncologists traditionally were treated with similarly intensive regimens regardless of whether they were adolescents, young adults, or middle-aged to older individuals. In the past 10 years, multiple retrospective comparisons of the outcomes of adolescents and young adults comparing those of similar age treated on pediatric-intensive regimens versus adult-intensive regimens have been made. Most of these studies have reported that survival outcomes are significantly better in adolescents and young adults (AYA) who receive therapy with pediatric regimens as compared with adult regimens. One of the first reports came from a review of data from the Children’s Cancer Group (CCG) in comparison with data from the Cancer and Leukemia Group B (CALGB). A comparison of 197 CCG patients with 124 CALGB patients showed that although the complete response (CR) rates were identical at 90% between the two groups, in the CCG study there was a 67% overall survival (OS) at 7 years compared with 46% for the CALGB cohort ( P <.001). For the CCG patients, central nervous system (CNS) prophylaxis was more intensive and given earlier in the course; moreover, doses of nonmyelosuppressive drugs such as asparaginase, vincristine, and corticosteroids were much higher in the CCG regimens than in the CALGB regimens. At least 6 other retrospective comparisons have largely demonstrated similar results, although the reader is cautioned that treatments in some of these studies were often quite different and incorporated blood and marrow transplantation (BMT) in different ways. There has been much speculation as to why patients treated on pediatric regimens have improved outcomes, and whether this relates to the higher doses of nonmyelosuppressive drugs, greater adherence to treatment schedules, and/or the greater experience of pediatric hematologists and oncologists in treating this disease.
These reports have led to the development of pediatric-intensive regimens for the treatment of adults. The French GRAALL (Group for Research and Adult Acute Lymphoblastic Leukemia) 2003 Trial was a phase 2 study in which a pediatric-inspired regimen was used to treat patients between the ages of 15 and 60 years. The regimen contained higher doses of prednisone, asparaginase, and vincristine in comparison with the group’s previous adult protocols. With a median follow-up of 42 months, the event-free survival (EFS) and OS were 55% and 60%, respectively. However, patients older than 45 years had a treatment-related death rate of 23% in contrast to a 5% rate in patients younger than 45. EFS in the younger patients was 58% versus 46% in the older patients ( P = .03). Other phase 2 studies have reported similar outcomes.
Randomized trials comparing pediatric-intensive with adult-intensive regimens in adults with ALL have not been reported, and may be logistically difficult to do. An alternative approach to addressing this question is being conducted in the United States through the Intergroup Trial led by CALGB, protocol 10403. This trial is taking patients with Philadelphia chromosome–negative (Ph-neg) up to the age of 40 years and treating them with one arm of a high-risk CCG protocol. The CALGB 10403 protocol will compare the outcomes of patients treated with this regimen with pediatric patients receiving therapy on a designated arm of the CCG protocol. End points for this study include use of traditional end points such as CR rates and survival, but will also assess whether patients and physicians are able to adhere to the scheduling and dosing requirements of the protocol.
Several new approaches in the therapy for ALL, including established agents used in other diseases and new agents in development, represent hope for therapeutic advances in the treatment of ALL and are outlined in detail in this review ( Box 1 ).
- •
Application of pediatric ALL regimens to AYA
- •
Monoclonal antibodies
- ○
Rituximab
- ○
Epratuzumab
- ○
CAT8015
- ○
CAT-3888
- ○
Inotuzumab ozogamicin
- ○
DT2219 ARL
- ○
Alemtuzumab
- ○
Blinatumomab
- ○
- •
Chemotherapy
- ○
NOTCH γ-secretase inhibitors
- ○
Forodesine
- ○
Decitabine
- ○
- •
Emerging Therapies
- ○
Methotrexate analogues
- ▪
Pemetrexed
- ▪
Talotrexin
- ▪
- ○
Flavopiridol
- ○
Proteasome inhibition
- ▪
Bortezomib
- ▪
- ○
Mammalian target of rapamycin inhibitors
- ▪
Rapamycin
- ▪
RAD001 (everolimus)
- ▪
CCI-779 (temsirolimus)
- ▪
- ○
MDM2 inhibitors
- ○
MEK/ERK inhibitors
- ○
PIM protein kinase inhibitors
- ○
Monoclonal antibody therapy
Immunophenotypic analysis of ALL plays a fundamental role in the diagnosis and classification of the disease. It is assuming an increasingly prominent role in the assessment of minimal residual disease in both pediatric and adult patients. A prominent antigen, CD20, is expressed on B-lineage lymphocytes in the mid-stages of development and is found on 40% to 50% of B-lineage ALL, with expression rates rising to 80% to 90% in mature B-cell or Burkitt-type leukemia and lymphoma. In pediatric ALL, expression of CD20 was found to be of conflicting prognostic significance with some studies suggesting inferior EFS, which was independent of other prognostic factors such as age and karyotype. Another study from St Jude Children’s Hospital suggested that expression of CD20 led to better outcomes. In adults, a retrospective analysis of 253 adult patients, of whom 47% expressed CD20 at a level of 20% or more, demonstrated that although CR rates were similar, disease recurrence was higher in the CD20-positive group. These patients had less lymphadenopathy but more thrombocytopenia and a poorer performance score than the CD20-negative patients. Remission duration and OS were poorer in the CD20-positive patients, and CD20 expression remained an independent predictor of outcome in multivariate analysis. The GRAALL Group has also reported that in Ph-negative B-lineage ALL in patients up to the age of 55 years, CD20 expression was associated with an increased risk of relapse (39% vs 20%, P = .02). Overall, it did not significantly affect EFS or OS (55% vs 59% at 42 months and CD20-positive and CD20-negative groups, respectively, P = .65). However, in patients with a white blood cell count of greater than 30 × 10 9 /L, EFS was much worse in the CD20-positive cohort (70% vs 24% at 42 months, P = .006).
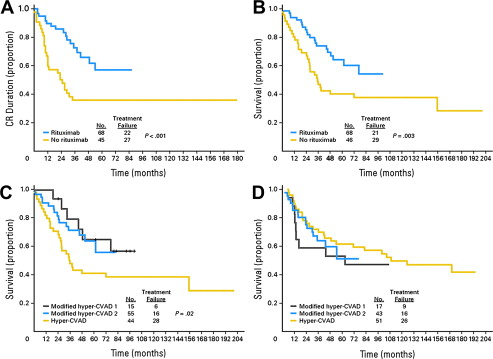
Rituximab, which has dramatically changed the therapy for non-Hodgkin lymphoma, has more recently been applied to therapy in CD20-positive B-lineage ALL. In a retrospective study of 282 adolescents and adults with de novo Ph-neg B-lineage ALL received a combination of hyper-CVAD plus rituximab versus a cohort who received hyper-CVAD alone. The CR duration and OS with hyper-CVAD combined with rituximab as compared with hyper-CVAD alone were significantly better. The CR duration was 70% with hyper-CVAD plus rituximab versus 38% with hyper-CVAD alone ( P <.001), and for OS 75% with hyper-CVAD plus rituximab versus 47% for hyper-CVAD alone ( P = .003) ( Fig. 1 ). These results were reported in patients younger than 60 years. Patients older than 60 years did not have any significant differences in outcome, whether or not they received rituximab. In a series of patients between the ages of 15 and 55 years from the German ALL Study Group, 41% of patients had CD20 expression of greater than 20%. In these patients, rituximab was added to the standard combination chemotherapy regimen. The CR rates were similar between the 181 patients who received rituximab plus chemotherapy versus the 82 patients who received chemotherapy alone. Early death rates were also similar. In standard-risk patients, achievement of minimum residual disease (MRD) negativity at day 24 was 57% with rituximab versus 24% without rituximab. Continuous CR at 5 years had a probability of 80% with rituximab versus 47% without rituximab, and overall survival was 71% with rituximab versus 57% without. For conventionally defined high-risk patients, the OS at 3 years was 55% with rituximab and 36% without rituximab. This improvement in outcome was thought to be attributable to fewer relapses in those who received rituximab. Of interest, CD20 expression may actually increase following exposure to corticosteroids, as demonstrated in a group of pediatric ALL patients in whom CD20 expression was upregulated from 45% in initial samples to 81% at the end of induction therapy. This result suggests that additional patients may benefit from rituximab. Rituximab is currently being studied in the therapy for newly diagnosed Ph-neg B-lineage ALL patients in phase 3 randomized trials.
Another antigen of interest in the therapy for ALL is CD22. This 135-kDa transmembrane sialoglycoprotein, a member of the immunoglobulin superfamily, is present in the cytoplasm of developing B cells. Cell surface expression occurs at a later stage of B-cell development. CD22 expression on B-cell malignancies is high, in the range of 60% to 80%. A CD22 IgG 1 humanized monoclonal antibody known as epratuzumab has been developed. It rapidly internalizes into the cell on binding to CD22, and appears to stimulate more of an immunomodulatory effect than an antiproliferative effect with rituximab. Epratuzumab has single-agent activity in aggressive B-cell non-Hodgkin lymphoma. It has been combined with rituximab and chemotherapy, demonstrating a high CR rate of 62% and a partial response (PR) rate of 33%, with EFS that appears superior to rituximab and chemotherapy alone.
The Children’s Oncology Group (COG) has evaluated epratuzumab in pediatric ALL patients in a pilot trial. Patients with leukemic blast expression of CD22 of greater than 25% received 4 doses of intravenous epratuzumab, 360 mg/m 2 per dose twice weekly for 4 doses and then the same dose weekly for 4 weeks in combination with chemotherapy. All patients were in marrow relapse and, of the 12 evaluable patients who received single-agent therapy, 2 patients had a minimal cytolytic response, 8 had stable disease, and 2 progressed. Nine of the 12 patients achieved a CR after combined epratuzumab and chemotherapy, for which 7 were negative by MRD. The COG is conducting a phase 2 trial of 4 doses of epratuzumab in combination with chemotherapy, and other trials combining epratuzumab with chemotherapy are ongoing.
Other CD22 antibodies of interest include a combination of an antibody that is fused to a 38-kDa fragment of Pseudomonas aeruginosa exotoxin A. This agent is known as CAT-3888. A phase 1 trial of 23 pediatric ALL patients with heavily relapsed disease gave doses ranging from 10 to 40 μg/kg every other day for 3 to 6 doses and repeated every 21 to 28 days. Adverse events were noted to be mild and reversible; and the trial did not demonstrate a maximum tolerated dose (MTD). Objective responses were not noted, but transient clinical activity was found. This immunotoxin has been modified to create higher CD22 binding affinity and is known as CAT-8015 or moxetumomab pasudotox. It has been tested in vitro and found to be highly cytotoxic to B-lineage ALL cells in preclinical studies. A humanized anti-CD22 monoclonal antibody has also been conjugated to calicheamicin, and is known as inotuzumab ozogamicin (CMC544). Studies in patients with refractory non-Hodgkin lymphoma have demonstrated an objective response rate of 87% in patients with follicular lymphoma and 80% in large B-cell lymphoma. In a phase 1 trial in patients with refractory and relapsed ALL of 36 evaluable patients, CD22 was expressed in more than 50% of the blasts in all patients and in more than 90% in 20 patients. Nine of 36 patients achieved a conventional CR, and an additional 11 had a marrow CR for an overall response rate of 56%. The agent was administered as an intravenous infusion over 1 hour every 3 weeks, and the median of courses administered was 2 (range 1–5). Fever and mild hypotension occurred in most patients on the first and second days of therapy. Nine patients or 25% had liver function abnormalities that were thought to be drug related, and these were severe in 4 patients. Two patients had liver biopsies showing periportal fibrosis. Twelve patients were able to proceed to allogeneic BMT, and 3 of these developed sinusoidal obstruction syndrome post transplant. Further trials using the drug in alternative schedules and in combination with other agents are planned.
An interesting approach to treating B-lineage ALL is to combine antibodies in a bispecific fashion. DT22199ARL is made up of two scFv ligands recognizing CD19 and CD22, which are linked to a hybrid gene encoding the first 390 amino acids of the catalytic diphtheria toxin (DT390). This molecule has been enhanced genetically so as to improve the in vivo antileukemic activity via reverse orientation of the VH-VL domains with the addition of linkers that reduce and stabilize aggregation. In an imaging model of a bioluminescent xenograft in which progression of a human Raji-Burkitt lymphoma cell line could be tracked, it was shown that this unique molecule could prevent hindlimb paralysis by blocking metastases to the spinal cord. This agent is given intravenously for 4 doses every other day for 4 hours on an inpatient basis, and is in the midst of phase 1 testing.
The anti-CD52 antibody, alemtuzumab (Campath-1H), is an unconjugated humanized monoclonal antibody. CD52 is expressed on virtually all normal and malignant T and B lymphocytes as well as monocytes and macrophages.
Experience with alemtuzumab in the treatment of ALL has been relatively limited. In a phase 2 study, 6 patients with ALL and 9 patients with relapsed-refractory acute myelogenous leukemia (AML) were treated with 30 mg intravenously 3 times a week for a total of 4 to 12 weeks. All patients demonstrated myelosuppression, but 10 patients progressed while on study, and there were frequent infections.
In a trial assessing the efficacy of alemtuzumab in eradicating MRD in ALL, the CALGB tested sequential cohorts of CD52-positive patients with a dose escalation of alemtuzumab to a target dose of 30 mg administered 3 times a week subcutaneously for 12 doses (4 weeks) during postremission therapy. Twenty-four patients ranging in age from 18 to 77 years were treated. The drug was generally well tolerated with mild hematologic toxicities, but grade 3 to 4 myelosuppression was evident. Two patients had transient viremia with cytomegalovirus. In 11 of 24 patients, serial assessment of MRD demonstrated a median 1-log decrease in disease burden. During subsequent postremission therapy, 8 patients developed cytomegalovirus viremia, 3 had herpes zoster reactivation, and 2 had herpes simplex infections. Further studies with a 30-mg dose are planned.
Another novel approach to using monoclonal antibody therapy in the treatment of ALL is to engage the cytotoxic benefit of T cells by constructing a bispecific T-cell engaging (BiTE) antibody construct. The agent, blinatumomab, combines the variable regions of an anti-CD19 antibody directed against B cells and an anti-CD3 antibody directed against T cells. This agent physically links T cells and malignant B cells, thus triggering the T-cell receptor complex signaling cascade. Recruitment of T cells with subsequent activation occurs when the anti-CD19 antibody binds its target antigen.
A phase 2 trial of blinatumomab at 60 μg/m 2 /d given by continuous intravenous infusion for 4 to 8 weeks demonstrated 7 PR and 4 CR for a total of 11 responses in 12 patients.
In the trials with lymphoma, it was noted that in patients treated with 15 μg/m 2 /d, dosing had bone marrow clearance of disease. This finding provided the rationale for using this dose by continuous infusion for 4 weeks in patients with B-lineage ALL. In a recently reported trial, patients in complete hematologic remission who had evidence of disease by MRD testing that was either persistent or recurrent were treated. Of 20 evaluable patients, 16 became MRD-negative after one 4-week cycle of therapy. Twelve of these 16 had been molecularly refractory to previous chemotherapy. The most common grade 3 or 4 toxicity was lymphopenia, with an incidence of 33%. Other adverse events included fever, chills, decrease of blood immunoglobulins, and hypokalemia. Most of these were transient. One patient had to permanently discontinue treatment because of a grade 3 seizure; this was reversible. Another patient had syncope with a convulsion. Blinatumomab appears to be a promising therapy in B-lineage ALL, and further trials in patients with MRD positivity, those with morphologic relapse, and those on front-line therapy with chemotherapy are planned.
Emerging therapies
NOTCH1 is a gene encoding a transmembrane receptor that is integrally involved in normal T-cell development and regulation. Although only rare cases demonstrate a chromosomal translocation involving this gene, it was found in 2004 that more than 50% of T-cell ALL (T-ALL) cases, including different immunophenotypic subtypes, have activating mutations in the NOTCH1 receptor involving the extracellular heterodimerization domain and/or the C-terminal PEST domain of NOTCH1 . The NOTCH1 receptor is a ligand-activating transcription factor, which transduces extracellular signals at the cell surface into alterations in gene expression in the nucleus. The receptor has a complex structure with 4 distinct subtypes involving the Delta and Serrate family of ligands. The receptor is a class I transmembrane glycoprotein that acts as a heterodimer ( Fig. 2 ). NOTCH1 is active at multiple stages of T-cell development. If NOTCH1 function is ablated, there is a complete block of T-cell development. Prognostically, NOTCH1 mutations are not associated with an unfavorable prognosis, and some series have suggested that it may confer a more favorable prognosis.
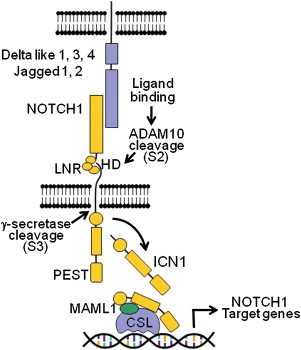
Given its high frequency, NOTCH1 would appear to be the suitable target for therapeutic maneuvers. There are several steps in the activation of NOTCH1 including processing by the γ-secretase complex, an aspartyl protease that cleaves NOTCH1 at the membrane level and releases intracellular components of NOTCH1 into the cytosol. By inhibiting the γ-secretase complex, it could be possible to prevent oncogenic NOTCH1 signaling in T-ALL. In Alzheimer disease, a presenilin γ-secretase complex plays an important role in disease pathogenesis. In Alzheimer disease, small-molecule γ-secretase inhibitors (GSI) block the generation of amyloidogenic Aβ peptides and may slow the progression of human dementia. Inhibitory molecules have been developed to block the processing of amyloidogenic Aβ peptides in Alzheimer disease. In vitro models showed that rapid clearance of intracellular activating NOTCH1 protein and transcriptional regulation of NOTCH1 target genes occurred with these agents. MK-0752, an oral GSI, was developed for the treatment of patients with relapsed ALL. In 7 T-ALL patients treated, 4 of whom showed a mutation in NOTCH1 , there were no objective clinical responses, and significant gastrointestinal (GI) toxicity and fatigue were seen. The GI toxicity was attributed to inhibition of NOTCH1 signaling in the gut. It may be that the GSI tested was ineffective in this setting because these agents appear to exert a cytostatic effect as their primary mechanism and exert little or no apoptosis. Subsequent studies with GSI have shown that they can reverse glucocorticoid resistance in T-ALL, and in vitro, corticosteroids inhibit the severe secretory metaplasia that is induced by inhibition of NOTCH1 signaling in the gut by GSI. Other GSI have shown promising activity in vitro and are in early clinical trials.
A genetic disorder leading to deficiency of purine nucleoside phosphorylase (PNP) results in specific T-cell immunodeficiency. This finding has suggested that inhibition of PNP could serve as a therapeutic pathway for treatment of T-ALL. The inhibition of PNP results in elevation of 2′-deoxyguanosine with accumulation of intracellular deoxyguanosine 5′-triphosphate. Accumulation of these substances induces cellular apoptosis. Bcx-1777, also known as forodesine, is a PNP inhibitor. For patients with relapsed or refractory T-ALL, forodesine was infused at a dose of 40 mg/m 2 intravenously 5 days per week for a total of 6 cycles. Dose escalations to 90 mg/m 2 were permitted in patients who were not responding after the second cycle. The overall response rate in 34 patients was 32%, with 7 CR and 4 PR. A BMT was able to be performed in 2 of the patients who achieved a CR. Dose escalation to 90 mg/m 2 after cycle 2 was performed in 18 patients. Cytopenias, nausea, headache, and asthma were the adverse events noted. Forodesine has also shown activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia.
Hypermethylation of densely clustered cytosines (CpG islands) within the promoter region of tumor suppressor genes has been shown to induce gene silencing in ALL. Exposure of ALL cell lines to decitabine has been shown to result in reactivation of epigenetically silenced genes and induction of apoptosis via global and gene-specific hypomethylation. The pyrimidine nucleoside analogue, decitabine, is an inhibitor of DNA methyltransferase activity and can reactivate silenced genes by inhibiting methylation of cytosine residues on DNA. A phase 1 trial of decitabine in relapsed and refractory ALL initiated therapy at 10 mg/m 2 for 5 days every other week. Dose levels ranging from 10 to 120 mg/m 2 were given to 23 patients with an overall response rate of 26%. There was 1 CR with incomplete platelet recovery (CRp) and 5 complete marrow responses of blasts under 5%. Toxicities included diarrhea, fatigue, and liver function test abnormalities. Responses were noted at multiple dose levels, and global hypomethylation was seen in vitro. A phase 1 trial with or without hyper-CVAD in ALL was recently reported. Patients with relapsed or refractory ALL qualified for the study without regard to the presence or absence of CNS disease, performance status, age, or level of hepatic or renal function. Patients were initially treated with single-agent decitabine, and those patients not responding or those that lost their response could be treated in the sequential phase of the trial with a combination of hyper-CVAD and decitabine. The initial dose of decitabine used was 10 mg/m 2 intravenously, daily for 5 days every 2 weeks as a single agent. When combined with hyper-CVAD, the initial dose was 5 mg/m 2 daily for 5 days on a 28-day cycle. In total, 39 patients were treated with a median age of 33 years (range 4–67 and a median white blood cell count of 5.8). Patients had received a median of 3 prior therapies (range 1–7). All patients but 9 had complex cytogenetics, with 4 of the remaining 9 having a Philadelphia chromosome. Single-agent decitabine dose levels ranged from 10 to 120 mg/m 2 /d for 5 days. Seven of 30 patients (23%) treated with single-agent decitabine achieved complete marrow responses. Responses were observed at all dose levels but were transient. A dose of 60 mg/m 2 intravenously, daily for 5 days every 14 days was selected as the optimal dose because of lack of toxicity and induction of global hypomethylation. Of the 30 patients treated with decitabine alone, 16 went on to receive decitabine and hyper-CVAD in a sequential fashion. An additional 9 patients went directly to therapy with decitabine and hyper-CVAD. Doses up to 60 mg/m 2 intravenously, daily for 5 days were combined with hyper-CVAD. No significant toxicity was associated with the combination therapy. With the combination, 13 of 25 patients (52%) achieved a response including 4 CR (16%), 2 CRp (8%), and 7 complete marrow responses (28%). Median duration of response was longer than 4 months. The optimal dose of decitabine in combination with hyper-CVAD was thought to be 40 mg/m 2 intravenously, daily for 5 days. The investigators concluded that decitabine has single-agent activity and in combination with hyper-CVAD is safe.
Folic acid antagonists such as the classic drug, methotrexate, were among the first agents to show activity in the treatment of ALL. A recently developed folate antagonist of interest is pemetrexed (LY-231514). Pemetrexed has been used in a phase 1 trial of patients with relapsed or refractory acute leukemia and has shown modest activity, with one patient with ALL achieving a PR at a dose level of 3.6 g/m 2 . Liver dysfunction was the main nonimmunologic adverse event. The recommended phase 2 dose was 2.7 g/m 2 over 25 minutes every 3 to 4 weeks with vitamin supplementation. It was thought that the drug had limited activity as a single agent, but study of its use in combination with other agents might be considered.
Another antifolate drug of interest is talotrexin, which has a higher affinity than methotrexate to dihydrofolate reductase. It achieved cell entry through a reduced folate carrier type 1 enzyme. Talotrexin has been administered as 5- to 10-minute infusion daily on days 1 to 5 over 5 to 10 minutes on a 21-day schedule, and a major response was noted in a patient with refractory ALL in an ongoing phase 1 study.
Another agent of interest is the synthetic flavone derivative, flavopiridol (Alvocidib), which is derived from tree bark. It has been shown to induce apoptosis and inhibit growth against multiple human tumor cell lines, and has activity in both myeloid and lymphoid malignancies. In a phase 1 trial as a single agent, a novel bolus/infusion schedule was used whereby the initial dose is given over 30 minutes followed by a second dose infused continuously over 4 hours. The MTD in a single-agent phase 1 study in relapsed and refractory AML and ALL was found to be 40 mg/m 2 by intravenous bolus followed by 60 mg/m 2 by continuous intravenous infusion. Secretory diarrhea was the dose-limiting toxicity. As a single agent, disease activity was modest. Dramatic but transient reductions in circulating blasts that lasted 10 to 14 days were seen. In a phase 1 trial in which flavopiridol was given as a daily 1-hour infusion for 3 days in combination with cytarabine and mitoxantrone to patients with refractory-relapsed AML or ALL, 40% of patients demonstrated direct leukemic cytotoxicity with a greater than 50% drop in peripheral blast counts and tumor lysis in 9 of a total of 34 patients. The overall response rate was 12.5% in the ALL patients, whereas it was 31% in 26 AML patients.
Bortezomib is the first agent in a class of drugs that inhibits the proteasome. This process represents a new mechanism for inducing apoptosis in malignant cells. Bortezomib has been extensively studied in multiple myeloma and relapsed non-Hodgkin lymphoma, treatment of which by these agents is approved by the Food and Drug Administration. Preclinical data suggest bortezomib might have single-agent activity in ALL. In a phase 1 trial with bortezomib in relapsed and refractory acute leukemia, the MTD was determined to be 1.25 mg/m 2 using a twice-weekly schedule for 4 out of 6 weeks. Proteasome inhibition was dose dependent and was 68% at the limiting dose of 1.5 mg/m 2 . Five of 15 patients showed evidence of hematologic improvement. In 12 pediatric patients (9 with ALL and 3 with AML), a phase 1 study was conducted, and the recommended phase 2 dose was suggested to be 1.3 mg/m 2 per dose given twice weekly for 2 weeks followed by 1 week of rest. Unfortunately, no objective responses were seen in this study. However, bortezomib has been combined with conventional ALL agents in children including doxorubicin, pegylated asparaginase, dexamethasone, and vincristine in a phase 1 study in which the bortezomib was dose escalated on days 1, 4, 8, and 11. In this combination, 6 CR were seen among 9 evaluable patients with a dose of bortezomib of 1.3 mg/m 2 .
A serine and threonine kinase, the mammalian target of rapamycin (mTOR) acts through the PI3K-AKT transduction pathway and regulates antiapoptotic and proliferative signals. Examples of agents that inhibit mTOR include the first-generation drug, sirolimus (rapamycin), and second-generation analogues such as everolimus (RAD001) and temsirolimus (CCI-779). Induction of apoptosis by rapamycin has been demonstrated in B-lineage ALL cell lines in vitro, and has been shown to have in vivo activity against B-precursor leukemia/lymphoma in transgenic mice.
An in vivo model of pediatric ALL has been used to demonstrate that everolimus induces autophagy in ALL cells. In this study, everolimus increased Beclin-1 expression with focal degradation of cytoplasmic areas sequestered by autophagic structures as demonstrated by electron microscopy. In this study, where nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice were inoculated with childhood B-cell ALL cells, everolimus increased the median survival of mice from 21 to 42 days. When combined with vincristine, they had significantly increased survival compared with either treatment alone. Everolimus induced cell cycle arrest in a G 0/1 phase, and reduced levels of cyclin-dependent kinases 4 and 6 and dephosphorylation of the retinoblastoma protein.
Murine double minute (MDM2) protein has been shown to be overexpressed in pediatric ALL. It is often associated with a wild-type p53 phenotype and resistance to chemotherapy. Inhibition of the MDM2-p53 interaction by nutlin-3 can induce p53 activity and result in apoptosis, as has been shown in adult T-cell leukemia cells. R05045337 (RG-7112) is an oral formulation of nutlin-3 that is in phase 1 testing, administered either daily for 10 days or every other day for 11 of every 28 days.
Mitogen-activated protein kinase (MEK) can enhance proliferation and survival in leukemic cells when activated. MEK inhibitors might be therapeutic in ALL. BCL-2 interacting mediator of cell death (BIM), a proapoptotic protein, is inactivated by extracellular signal-regulated kinase (ERK)-mediated phosphorylation. In ALL cells, BIM is upregulated by dexamethasone. The use of MEK-ERK inhibitors in combination with dexamethasone may induce synergistic enhancement of apoptosis in vitro. Several MEK inhibitors are in clinical trials. The serine-threonine PIM kinases are upregulated in several hematologic malignancies and play important roles in multiple signal transduction pathways. T-lymphoblastic leukemia/lymphoma cells have been found to be sensitive to small-molecule inhibitors of PIM protein kinases in vitro, and in vivo in immunodeficient mice inoculated with these malignant cells.
Related posts:
 Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Prognostic Factors in Adult Acute Leukemia
Oddballs: Acute Leukemias of Mixed Phenotype and Ambiguous Origin
Acute Leukemias
Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Novel Transplant Strategies in Adults with Acute Leukemia
Induction and Postremission Strategies in Acute Myeloid Leukemia: State of the Art and Future Directions
Prognostic Factors in Adult Acute Leukemia
Oddballs: Acute Leukemias of Mixed Phenotype and Ambiguous Origin
Acute Leukemias
Clinical Implications of Novel Mutations in Epigenetic Modifiers in AML
Novel Transplant Strategies in Adults with Acute Leukemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


