Gene
Disease
Clinical features
Prevalence amongst MODY patients
Therapy
1
Hepatocyte nuclear factor-4-alpha 20q
MODY 1
Macrosomia, neonatal hypoglycaemia and hyperinsulinaemia; after puberty insulin deficit and diabetes [2]
Rare
SUL.
2
Glucokinase 7p
MODY 2
Childhood, young adult. Mild fasting (>100 mg/dl) and/or postprandial hyperglycaemia (>140 mg/dl)
Glycaemia at time 120′ of OGTT
Glycaemia at time 0′ < 50 mg/dl Slight elevation of HbA1c
Common
None
3
Hepatocyte nuclear factor-1-alpha 12q
MODY 3
Severe and progressive hyperglycaemia at time of puberty or in young adults, with tendency to ketoacidosis. Low renal threshold for glucose. Possibility of microvascular complications. The volume of the exocrine pancreas is reduced [5]
Common
SUL.
4
IPF-1 13q12.1 pancreas/duodenum homeobox protein-1 gene
MODY 4
Homozygous (neonatal) = pancreatic agenesis
Heterozygous = mild diabetes with onset in adult [6]
Rare
Diet, OHA, INS.
5
Hepatocyte nuclear factor-1-beta 17q
MODY 5
Rare
INS.
6
NEURO D1 2q32
MODY 6
Mild diabetes in young adult. Low serum insulin levels
Very rare
Diet, INS.
7
KLF-11 2p25
MODY 7
Fasting hyperglycaemia, mild diabetes in young adult [9]
Very rare
Diet, OHA, INS.
8
CEL 9q34
MODY 8
Young adult. Diabetes-pancreatic exocrine dysfunction syndrome [10]
Very rare
DIET, OHA, INS.
9
PAX4Chr 7q32
MODY 9
Diabetes in young adult. Only patients described from Thailand [11]
Very rare
Diet, OHA, INS.
10
INS 11p15.5
MODY 10
Childhood. C-peptide low and insulin high before the onset of the treatment [12]
Very rare
INS.
11
BLK 8p23
MODY 11
Mild diabetes [13]
Very rare
Diet, INS.
12
SUR 11p15.1
PNDM
TNDM
Neonatal diabetes, possible growth delay [14]
Rare
SUL.
13
KIR6.1 11p15.1
PNDM
TNDM
Neonatal diabetes, sometimes neurological disorders [15]
Moderate
SUL.
In order to understand the pathogenic mechanism of these mutations, it is useful to remind the physiology of the pancreatic beta cell’s secretion (Fig. 12.1).
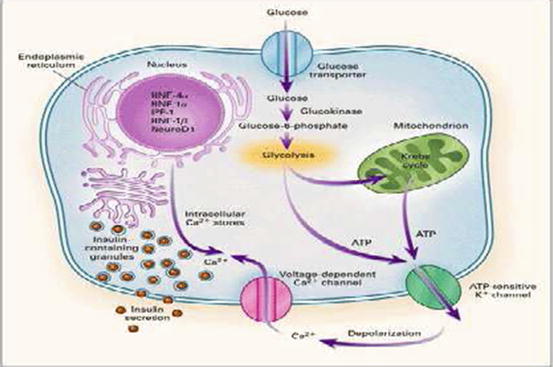
Fig. 12.1
Glucose enters the beta cell, proportionally to glycaemia levels, through Glut 2 receptor and is attached by glucokinase and transformed in glucose 6 phosphate in the glycolysis, and in the mitochondrion, ATP is produced; ATP closes the beta cell’s potassium channels (K-ATP) so the potassium accumulated inside the cell causes the depolarization of its membrane and the opening of the calcium channels that let the calcium enter, releasing insulin proportionally to the amount of the glucose that entered the beta cells [5]
Mutations in genes involved in insulin secretion or in nuclear beta-cell transcription factors that limit the ability of the pancreas to produce insulin are the cause of the major non-autoimmune diabetes forms of the paediatric age.
Nowadays 13 forms of MODY have been described. The term MODY (maturity-onset diabetes of the young) relies on the old classification of diabetes into juvenile-onset and maturity-onset diabetes.
In the 1970s Tattersall and Fajans named MODY the onset of an ‘adult diabetes’ even before the age of 25, with autosomal dominant transmission [5]. A revised, aetiology-based classification for diabetes was introduced by both the American Diabetes Association and the World Health Organization, and MODY is now included in the group of ‘genetic defects in beta-cell function’ with a subclassification according to the gene involved GCK-MODY, HNF1alpha-MODY, etc [7]. In addition to these monogenic diabetes forms, other hereditary diabetes forms have been recognised and characterised that develop usually in the first 9 months of life (even if there are cases in children and adolescents), in particular mutations in either KCNJ11 or ABCC8 genes, that code for Kir 6.2 and SUR1 subunits, respectively, of the pancreatic beta-cell K-ATP channel and the mutation of INS, the insulin’s gene [8].
The monogenic diabetes accounts for 1–2 % of all cases of diabetes in paediatric-adolescent age, but the apparent increasing in its prevalence is probably due to the improvement of the diagnostic methods [16].
Mitochondrial diabetes and type 2 diabetes of adolescents have been also described.
12.2 GCK-MODY (MODY 2)
GCK-MODY is an autosomal dominant inherited disease caused by heterozygous, inactivating mutation in the glucokinase gene (GCK), which encodes GCK enzyme. GCK acts as the pancreatic glucose sensor, and the heterozygous loss-of-function mutations cause an increased threshold for the initiation of glucose-stimulated insulin secretion [17].
This leads to mild and stable hyperglycaemia, with fasting plasma glucose values of 5.5–8 mmol/l, since birth. Furthermore, GCK-mutated patients show a small increment of glycaemia at 2 h (<4.5 mmol/l) during an OGTT.
As a result, GCK-MODY patients have HbA1c levels just above of upper limit of normal and rarely higher than 7.5 % (58 mmoli/moli). This explains why carriers of GCK mutations are usually not at risk to develop micro- or macro-vascular complications and, with rare exceptions, do not require pharmacological treatment [18, 19].
Patients are generally asymptomatic, and hyperglycaemia is commonly found out incidentally. For this reason a familial history of diabetes in a first-degree relative may be not known, and testing apparently unaffected parents for fasting glucose can be important when approaching a patient with mild fasting hyperglycaemia (rare de novo mutations have been described). Thus, GCK-MODY can be diagnosed at any age.
Noteworthy is that the age of diabetes detection in GCK-MODY patients will influence the clinical classification and therapeutic choices of diabetologists. In fact, individuals can be misdiagnosed with type 1 diabetes and treated with insulin, if detected during childhood, or with type 2 or gestational diabetes, if detected in adulthood or in pregnancy, respectively [20].
In particular, the right diagnosis of GCK-MODY in pregnancy is very important as 50 % of offspring is likely to inherit the GCK mutation and that will influence the foetal size. Foetus who does not inherit the maternal mutation has an increased risk of macrosomia and postnatal hypoglycaemia, secondary to the maternal hyperglycaemia. For these reasons, insulin is required in cases of excessive foetal growth. Conversely, insulin treatment of GCK-mutated woman where the foetus is affected may result in low birth weight [21].
The exact prevalence of GCK-MODY is unknown, and it is estimated between 30 and 60 % of all MODY cases according to the geographic area and the clinical setting [22]. GCK mutations represent the commonest MODY defect in the paediatric setting in Italy [3, 23].
Genetic testing is the gold standard for diagnosing monogenic diabetes, but it is still expensive and not easily accessible. Hence, a proper clinical selection of the candidate for the molecular testing according to European Molecular Genetics Quality Network (EMGQN) guidelines is necessary [18].
However, not all clinicians have the expertise to recognise the clinical features of monogenic diabetes. Therefore, we designed and validated a 7-item flowchart (7-iF) to identify patients with a high risk to carry a pathological GCK mutation, improving the detection rate and reducing the number of unnecessary tests. The 7-iF consists of seven questions with binary answers (yes/no) about clinical, biochemical and anamnestic data (Table 12.2). This study shows that a score ≥6 should be considered an indication for the genetic test. Nevertheless, the patients with a score less than 6 need to be referred to a specialised centre in order to spot the few patients who might be still affected by monogenic diabetes [24].
Table 12.2
Seven items flowchart (7IF)
Indications to GCK-MODY 2 molecular tests | |
|---|---|
Disclaimer statement: Results obtained from the proposed questionnaire should never replace clinician’s advice and opinion. The final decision about any diagnostic and therapeutic procedure is under the clinicians’ responsibility | |
1. Absence of autoimmune markers | |
Answer | Indication for test |
Negative marker | Yes |
At least one marker positive | No |
Markers not checked | No |
2. Current or past insulin therapy | |
Answer | Indication for test |
No insulin therapy | Yes |
Insulin therapy | No |
3. HbA1c values | |
Answer | Indication for test |
HbA1c values ≥42 mmol/L (6 %) | Yes |
HbA1c values <42 mmol/L (6 %) | No |
4. Onset (diabetes or hyperglycaemia) >6 months or <25 years | |
Answer | Indication for test |
After 6 months and before 25 years | Yes |
Before 6 months of age | No |
After 25 years of age | No |
5. Positive familiarity for diabetes, IFG or IGT | |
Answer | Indication for test |
At least one parent affected | Yes |
No affected parent | No |
6. Signs of different type of diabetes | |
Answer | Indication for test |
None | Yes |
Obesity (BMI > 30 kg/m2 in adults or z-score >2 in children)
Related posts: Hvidoere Study Group: What Can Be Learned from Observational Studies
Long-Term Microvascular Complications: New Ideas for Research
Nutritional Aspects of Type 1 Diabetes: We Need to Keep Struggling Against Palaeolithic Diet (How Research Helps Us to Do the Right Thing) Hvidoere Study Group: What Can Be Learned from Observational Studies
Long-Term Microvascular Complications: New Ideas for Research
Nutritional Aspects of Type 1 Diabetes: We Need to Keep Struggling Against Palaeolithic Diet (How Research Helps Us to Do the Right Thing)
 Insulin and Immunotherapy in Children and Adolescents with Type 1 Diabetes Insulin and Immunotherapy in Children and Adolescents with Type 1 Diabetes
 Glucose Sensors Glucose Sensors
 Advanced Pump Functions: Bolus Calculator, Bolus Types, and Temporary Basal Rates Advanced Pump Functions: Bolus Calculator, Bolus Types, and Temporary Basal Rates
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |