Chapter 60 Non-Hodgkin Lymphoma
INTRODUCTION
Non-Hodgkin lymphoma (NHL) is a broad term encompassing many neoplasms of lymphocytes, and along with Hodgkin lymphoma (HL) is the most commonly occurring hematologic malignancy in the United States.1 Compared to the general population, the incidence of NHL and HL in persons with human immunodeficiency virus (HIV) infection is markedly elevated.2 In 1985 the US Centers for Disease Control (CDC) included biopsy-confirmed NHL of high-grade pathologic type (diffuse, undifferentiated) and of B-cell or unknown immunologic phenotype, in the setting of HIV infection, as a case definition for acquired immunodeficiency syndrome (AIDS).3 AIDS-related lymphoma (ARL) is one of the most lethal complications of HIV infection,4 although its prognosis has improved since the introduction of highly active antiretroviral therapy (HAART).5,6
The prognosis for ARL is worse than that for similar lymphomas occurring in non-AIDS patients.6–8 Recently, HAART has been associated with changes in the epidemiology and natural history of these tumors. Therapeutic approaches have become more similar to non-HIV NHL. Pre-HAART, the therapeutic goal was frequently palliation, since the prospects for long-term survival were generally poor. In the HAART era, certain subsets of patients with AIDS and lymphoma may have outcomes equivalent to those of their counterparts in the general population.9,10 Consequently, curative intent treatment is appropriate for many, if not most, patients with ARL. Lymphoma cure depends on achieving a complete remission of the tumor without subsequent recurrence, and recurrence is a major cause of treatment failure in lymphoma. Identification of patients most likely to have a favorable outcome is informed on the basis of the underlying AIDS and appropriate tumor classification. Optimal treatment planning must take these features into account.
NHL encompasses a broad spectrum of lymphocyte neoplastic diseases. In non-AIDS patients, optimal management and prognosis are dependent on the specific lymphoma type. In ARL, the specific lymphoma type has until recently been less relevant to either treatment or to prognosis. Lymphoma classification incorporates morphologic, immunophenotypic, and clinical features. Recent advances in identifying the presumed cell of origin from which specific lymphoma types derive have helped in unraveling the underlying biology of various lymphomas and distinguishing various lymphoma subtypes. This information helps place into perspective the finding that while lymphomas occurring in AIDS share similarities with HIV-unrelated lymphomas, they also have distinguishing epidemiologic and clinical features (Table 60-1).
Table 60-1 Characteristics of Peripheral Aggressive Lymphoma in AIDS and Non-AIDS
| Characteristic | AIDS | Non-AIDS |
|---|---|---|
| % of cancers | 20–30% | 5% |
| Proportion of all NHL with aggressive histologic features | 90% | 50% |
| High grade | 75% | 10% |
| Extranodal involvement | 40–80% | 40% |
| Leptomeningeal involvement | 17% | 10% |
| % NHL primary brain | 20% (pre-HAART) | 2% |
| ‘B’ symptoms | ≥80% | <30% |
| Age at onset | Second to third decade | Fifth to sixth decade |
From Clifford et al,20 National Cancer Institute,35 Biggar,158 and Jones et al.59
There is increasing evidence that in the HAART era, consolidation of ARL into a ‘catch-all’ quasidiagnostic label may be less useful than in the pre-HAART era. Also, it appears that lymphoma-specific disease features are of relatively greater prognostic importance than in the pre-HAART era.5,10 In order to confirm this early observation, inclusion of appropriate diagnostic refinement and subset analysis in clinical trails of ARL will be essential. In non-AIDS, outcome is critically dependent on appropriate histology guided treatment. In the HAART era, this may prove to be the case for ARL.
The World Health Organization Classification of Tumors has broadened the original CDC spectrum of lymphomas associated with HIV infection, as listed in Table 60-2. These lymphomas can have markedly different clinical characteristics and prognosis, and understanding these differences is essential toward optimal management, clinical trial development, and disease surveillance.
Table 60-2 Categories of HIV-Associated Lymphoma
From Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). World Health Organization Classification of Tumors: Pathology & Genetics: Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001.
EPIDEMIOLOGY
Early after AIDS was recognized as a new disease, it was apparent that NHL was one of the complicating opportunistic illnesses. In contrast to AIDS-related Kaposi sarcoma (KS) which occurred with greatest excess among men who had sex with other men (MSM), the risk of NHL appeared to be unrelated to the HIV risk acquisition factors.2,11 In the pediatric population, the incident cases were ∼300-fold greater than observed in general population, although since NHL is a rare pediatric disease, there were not a great number of cases.2 However, in young adults aged 20–40 years, the incident cases were found to be over 70-fold greater than expected, leading to ∼3000 cases by 1989 compared to the ∼50 cases expected for the period. In addition, although not an AIDS-defining condition, it became clear that the incidence of HL was increased. There is an eight- to 10-fold increase of HL in HIV-infected compared to the noninfected population. HL is the most common non-AIDS-defining cancer among individuals with HIV infection.12,13
Epidemiologic features of ARL have been modified by the introduction of HAART. Most studies have shown ARL to have decreased in incidence by ∼50% since HAART,6,14 although the proportion of AIDS-defining illness (ADI) that is due to lymphoma appears to be somewhat increased, owing to the relatively greater decrease in other ADIs.15,16 Also, lymphoma is now nearly equal to or exceeds KS as the leading malignant AIDS-defining event.17–20 Where HAART is readily available, cancer has overall become the leading cause of death in HIV-infected persons, and NHL is the leading cause of cancer-related death in AIDS.21,22
The risk of developing ARL is inversely related to CD4+ cell count and in populations where HAART is widely used, the proportion of individuals with low CD4+ cells is reduced, resulting in a decrease in ARL incidence.20 Also, the use of HAART has resulted in a change in the relative distribution of ARL subtypes. This appears to be explained in part by the observation that there is a correlation between CD4+ cell count and the risk of developing a specific ARL type.17,23 Most studies show that only those lymphoma subtypes that are most associated with advanced immune depletion are particularly decreased in incidence in the HAART era, though there is no complete agreement in the data.5,6,17
The greatest decrease in incidence has been seen in the immunoblastic variant of diffuse large B-cell lymphomas (DLBCL), a subtype strongly associated with advanced CD4+ cell count depletion and Epstein–Barr virus (EBV) infection. This variant accounted for ∼60% of all ARL prior to HAART,2 and has substantially decreased in incidence so that since HAART was introduced, the immunoblastic variant represents ∼30% of cases.17 However, caution should be taken in the interpretation of such data because there is no uniform agreement among pathologists as to what establishes the diagnosis of immunoblastic lymphoma. Prior to HAART, AIDS-related primary central nervous system lymphomas (AR-PCNSLs), which are virtually all of the immunoblastic variant, accounted for ∼20% of all ARLs.2 Since the introduction of HAART, tumor has decreased in incidence by ∼70%.6 Interestingly, this subtype is nearly always EBV associated.
In a large retrospective review, Besson and colleagues reported a statistically nonsignificant increase in AIDS-related B-cell lymphoma (AR-BL) in the HAART era from 17.7% to 26.8% of cases.6 DLBCL cases were decreased from 79% to 63.4% of overall ARL cases. A notable finding from these data is that within CD4+ cell strata, no overall change in ARL incidence was seen, suggesting that the overall decrease in ARL incidence with HAART is due to a decreased population of individuals with low CD4+ cell counts. However, there is evidence that factors in addition to absolute CD4+ cell counts are likely involved in the complex changes in incidence of various lymphoma subtypes. As yet, with only less than one decade of data available for analysis, it may be too soon to fully appreciate the effects of HAART on tumor epidemiology in HIV infection.
While most data show that the incidence of the centroblastic DLBCL variant and Burkitt lymphoma (BL) has remained relatively stable since HAART was introduced,17,24 some studies report an increase in DLBCL and a decrease in Burkitt.5 Lim and colleagues identified a shift in the ratio of cases favoring the more likely occurrence of DLBCL over other subtypes by a factor of ∼3.5 in the HAART era in Southern California. This result is somewhat unexpected as DLBCL occurs with lower CD4+ cell counts than the BLs, and this result illustrates the complexity of the epidemiology and HAART effect.5 These interesting data underscore the uncertainly at present regarding the epidemiology in ARL, and also highlights the importance of rigorous lymphoma subset and variant classification.
Unlike EBV-associated ARL, which occur most frequently in the setting of low CD4+ cell counts, HIV HL tends to occur while the CD4+ cells are relatively intact, even though the vast majority are also EBV associated.25 In contrast to the EBV-associated NHL, HL appears not to have decreased, and if anything to have increased in incidence in the HAART era.17,20 Such epidemiological discrepancies underscore the differences in tumor pathophysiology, the role of EBV in HL and NHL, and the complex interaction between the immune system and viral oncogenesis.
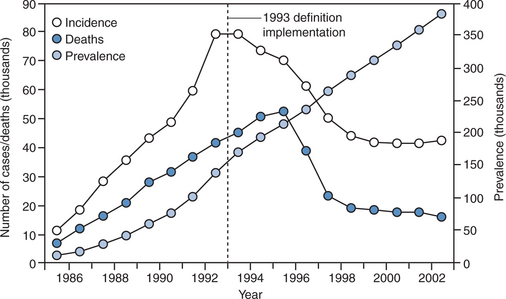
It remains to be seen how the overall burden of cancer in HIV-infected individuals will be affected by HAART. Although the risk of ARL and other AIDS-defining malignancies per patient-year has decreased with HAART, the at-risk population of persons with AIDS in the United States has grown by more than 50% since the introduction of HAART (Fig. 60-1). Since as yet no person has been cured of HIV infection, but treatment advances have translated into substantially increased longevity, this trend may be predicted to continue. Consequently, the prevalence of persons living with HIV/AIDS is likely to increase further. Moreover, as these individuals age, there may be an increased risk of developing NHL as is the case in the general population. It is conceivable that the future will bring new challenges to the medical oncology community and their infectious disease consultants if the absolute number of HIV-infected individuals with cancer increases commensurate with the already documented increase in AIDS prevalence.26
PATHOGENESIS
Lymphoma pathogenesis is a complex process that has not been fully unraveled. Clues to lymphomagenesis can be found from a synthesis of the various data available from epidemiologic, histologic, genetic, and molecular studies. For example, it has long been established that patients with congenital or acquired immune dysfunction have an increased risk of developing lymphoma, and that these are frequently viral associated.27 Additionally, pathogenic mechanisms have been informed by advances in the ability to identify the putative normal cellular counterpart from which the various lymphoma types derive. As the normal lymphocyte transits through the lymph node, the specific immune microenvironment is reflected by cell surface markers. Identification of such markers can be used to postulate the point when malignant transformation took place relative to passage through the nodal germinal center. This ontological information has helped to segregate lymphoma subsets into biologically and clinically relevant distinct entities.28,29 The specific histogenic pathways give rise to distinct biological and clinical patterns specific to the ARL subtypes (Table 60-3).30–32 Modification of the host immune microenvironment likely affects histogenesis, in part explaining the changing epidemiologic and clinical patterns of ARL in the HAART era.33
Over 80% of the NHL cases in HIV-infected patients are aggressive B-cell lymphomas,34 in contrast to the HIV-unrelated cases where less than 15% are of this type (Table 60-1).35 ARL are frequently associated with oncogenic gamma-herpesviruses, as is commonly the case with other immunosuppression-related lymphoproliferative disorders.26,27 Moreover, those ARLs associated with advanced immune depletion are more likely to be viral associated, whereas those that occur with higher CD4+ counts are less likely to be viral associated.30,33,36–38 As mentioned above, an exception to this is HL, which typically occurs prior to AIDS onset, and yet 80% or more are EBV associated.25,39,40 Also, it is possible that as yet undiscovered viral associations may be involved for those ARLs not currently known to be associated with oncogenic viruses. For example, the putatively oncogenic Simian virus 40 (SV40) has reportedly been found in association with some ARLs,41 although epidemiological analysis has not supported this as an etiologic agent in NHL.42
The association with gamma-herpesvirus infection in ARL invokes the possibility that these are lymphoproliferations that develop as immune control against the virus is lost. However, immunosuppression alone does not adequately explain why lymphoma is increased in HIV infection. Complex molecular events are involved in lymphomagenesis and in many cases, a series of events must occur for lymphomas to develop. The immunosuppression of AIDS may act in part to permit expansion of virally infected cells, especially those that express immunogenic markers, and thus increase the chance of subsequent genetic events that lead to frank lymphoma. As one example, EBV appears to rescue from apoptosis germinal center B-cells that have otherwise accumulated fatal somatic hypermutations.43 EBV proteins such as LMP1 and LMP2A can mimic the function of the B-cell receptors CD40 and BCR that are vital to cell survival. EBV may transform cells in part by overcoming loss of these cell proteins, providing a major role of the virus in the pathogenesis of HL and posttransplantation lymphomas.44 Such transformed cells also have an increased chance of developing other abnormalities without being destroyed by apoptosis.
Other factors that have been invoked in AIDS lymphomagenesis include chronic immune stimulation and cytokine dysregulation associated with HIV infection, and the response to these disturbances is in part dependent on innate host immune characteristics. For example, chemokines that are involved in normal B-lymphocyte maturation and proliferation are often dysregulated in patients with HIV. Also, certain gene variants of the chemokine receptor CCR5 and the CXC chemokine ligand-12 (stromal cell-derived factor-1) affect the risk of NHL in AIDS, independently of any potential protection from HIV itself.45,46 The finding that receptor variants affect lymphoma risk is consistent with the concept that HAART modification of immune dysregulation can through this mechanism affect lymphomagenesis and ARL epidemiology. A number of additional considerations support the role of immune microenvironment on ARL.
Since the subtype of ARL is dependent on histogenic origin, factors that influence B-cell transit through the lymph node are important to tumor biology.30,31 This provides another mechanism by which HIV can affect lymphoma histogenesis. As HIV disease advances, lymph node architecture is substantially altered.47 The nodal architecture to some degree reflects the effect of HIV on shortened T-cell survival and consequent changes in B-cell function.48 Also, the stage of HIV infection influences cytokine levels and these in turn are associated with the relative composition of germinal center cells and mature B-cells within the nodes and periphery.49,50 Thus, immune status, B-cell migration through the germinal center, and histogenesis may be intrinsically related. HAART can delay progression of HIV disease, and thus preserve nodal architecture, potentially influencing these relationships as reflected in the changing patterns of ARL epidemiology.
Gene expression profiling using DNA array technology provides strong evidence supporting the role of immune environment on ARL biology and epidemiology. Approximately 50–70% of ARL are DLBCL.5,51–53 Gene expression profiling has identified at least three major ontogenetically distinct DLBCL subtypes in non-AIDS.54 These subtypes have distinct tumor biology and clinical behavior. AIDS DLBCLs have similar gene expression patterns, except that a higher proportion of AIDS cases express the T-cell leukemia-1 proto-oncogene.55 Disruption of the immune microenvironment promotes expression of this oncogene.56,57 The increased incidence and aggressiveness of DLBCL in AIDS may thus be due to impaired immunosurveillance affecting expression of specific genes rather than changes in the overall gene expression pattern associated with prognosis of the various DLBCL subtypes. The decreased risk of developing ARL and improvements in ARL prognosis with HAART may in turn be related to changes in the expression of this oncogene. Whether expression of this oncogene influences development of specific DLBCL subtypes is not known.
Further evidence that the immune microenvironment influences tumor biology is the finding that the expression pattern of oncogenic viruses such as EBV can be modified by cytokines relevant to HIV disease stage. For example, interleukin-10 can induce the expression of LMP-1, demonstrating that this major transforming gene can be induced by extracellular signals.58 As HIV disease progresses, T-helper type II (THII) cytokines such as IL-10 become elevated,59 potentially promoting viral oncogenesis through its effects on viral latency patterns and giving rise to those tumors more likely to be associated with poor prognosis.30,31
Additional evidence that HAART has influenced ARL histogenesis comes from studies of the BCL-2 antiapoptotic protein. BCL-2 is one of the pathobiological markers predictive of poor prognosis in NHL, and appears to track with DLBCL arising from nongerminal center B-cells.29,60–63 Some evidence suggests that since HAART was introduced, fewer ARL over-express BCL-2 and that this may partially explain the improved prognosis, though not all data agree on this.8,9 Thus, while several lines of evidence support the notion that HAART has improved ARL prognosis through its effect on tumor histogenesis and subsequent tumor biology, this concept is not proven. As mentioned above, AR-PCNSL, which is universally EBV-associated and associated with profound immune depletion, rarely occurs with successful HAART. Table 60-3 summarizes the major pathobiological and viral markers that distinguish the chief ARL subtypes and immune correlates.
The discovery in 1993 of the KS-associated herpes virus (KSHV, also known as human herpes virus-8 or HHV-8),64 has further informed the relationship between viral oncogenesis and immune function. KSHV is the etiologic agent of primary effusion lymphoma (PEL), described later in this chapter. These lymphomas appear to derive from postgerminal center preterminally differentiated B-cells with an absence of the other genetic lesions commonly found in other ARLs, a feature associated with advanced immune depletion (Table 60-3).30,65,66 KSHV appears to be a causative oncogenic virus in these tumors, which primarily involve the body cavities as lymphomatous effusions.67 The particular association of this tumor with HIV infection and its presentation in the body cavities may in part be explained by a number of interesting properties of KSHV (Fig. 60-2). HIV can promote KSHV infectivity of new cells.68 KSHV encodes for hypoxia response elements, and can be activated by hypoxia. The virus may then enter lytic replication when an infected cell enters body cavities, which are relatively hypoxic.69,70 This environmental stimulus could potentially result in activation of lytically expressed KSHV oncogenes including viral BCL-2 (vBCL-2), and proangiogenic factors such as viral interleukin-6 and others.69–73 The KSHV-encoded G protein-coupled receptor, which is constitutively active, upregulates vascular endothelial growth factor perhaps contributing to the characteristic accumulation of body cavity effusions, thus further promoting the hypoxic environment that may be conducive to this tumor’s relatively niche-specific growth. Since PEL can present occasionally as solid lymphoma, these mechanisms do not fully explain the tumor biology, and other mechanisms are also important.
Plasmablastic lymphoma of the oral cavity, initially described as a rapidly growing tumor localized to the oral cavity or the jaw, is typically seen only in HIV-infected patients.74 Morphologically these are similar to DLBCL, but do not express CD20 and expression of the leukocyte common antigen is minimal or absent. The tumor cells react with the plasma cell characteristic antibody VS38c and also frequently with the CD79a antibody, both B-cell markers. They variably express cytoplasmic immunoglobulin and a monoclonal rearrangement of the immunoglobulin heavy chain gene, confirming the clonal B-cell origin, and the plasmacellular differentiation of these neoplasms. Most are EBV associated. B-cell histogenic markers confirm a postgerminal center origin,31 consistent with the poor prognosis the diagnosis generally implies. In the HAART era, there is evidence of improved outcome and that diffuse involvement of the lymphoma in multiple sites and organs may occur.75–78
HL in HIV (HIV-HL) has several biologic and clinical characteristics distinguishing it from classical HL in the general population.25,79 HIV-HL is EBV associated in 80% or more of cases, whereas in the general population less than 50% of cases are EBV associated. HIV-HL is most often of the mixed cellularity type without substantial mediastinal involvement, whereas in the general population HL is most often the nodular sclerosis histologic subtype, and often presents with bulky mediastinal enlargement. HIV-HL is more likely to present with advanced stage disease in younger patients than expected in HL occurring in the general population, and to be more likely associated with extranodal involvement and ‘B’ symptoms (unexplained weight loss, fevers, and drenching night sweats).40 The Reed–Sternberg cells of HIV-HL appear to derive from postgerminal center B-cells and although they express CD40, they are not surrounded by CD40 ligand-positive (CD40L+) reactive T lymphocytes, which are thought to regulate the disease phenotype through CD40/CD40L interactions in cases occurring outside HIV.80 The EBV-encoded LMP-1, which being functionally homologous to CD40, may contribute in part to the modulation of the HIV-HL phenotype.
PERIPHERAL ARL: CLINICAL FEATURES
General
ARL is a serious complication of AIDS and is associated with poorer survival than similar lymphomas in the general population.6,7,81 Moreover, ARL is one of the more rapidly lethal of AIDS-defining events, though the prognosis of certain histologic types has substantially improved in the HAART era.4,5 Since the introduction of HAART, median ARL survival has improved from 2–11 months to ∼2 years, but this does not extend to all ARL subtypes.5,6,82,83 Some small studies have shown AR-DLBCL progression-free survival to be about equivalent to non-AIDS lymphoma counterparts.9 Emphasizing the emerging importance of histologic subtype in ARL, Lim and colleagues found that improved survival since HAART is restricted to DLBCL and that Burkitt prognosis remains poor since the advent of HAART.5 The median survival of patients with DLBCL in the HAART era appears to be greater than 40 months compared to less than 6–18 months pre-HAART, whereas median survival for Burkitt remains at ∼6 months across eras.5,82 This finding potentially highlights that in the HAART era, lymphoma-specific disease features have become more important.
Presentation, Diagnosis, Staging
More advanced cases can present with bulky adenopathy, either as solitary or diffusely involved anatomic sites, and often including extranodal sites. The presenting symptoms and signs are related to the anatomic location of tumor involvement, and virtually any anatomic location can be involved, a feature that distinguishes ARL from the majority of non-AIDS NHL cases. Elevations in the serum lactate dehydrogenase (LDH) are often found in aggressive lymphoma, and have prognostic implications.84 Patients can present with severe constitutional symptoms referred to as ‘B’ symptoms characterized by drenching night sweats, unexplained fevers, and unexplained weight loss in excess of 10% of the body weight.85
Early in the AIDS epidemic, the prognosis for ARL was often very poor, and early diagnosis did not generally confer a clinical advantage. Consequently, aggressive diagnostic maneuvers were not always appropriate. With current treatments and improved outcomes, a more aggressive search for lymphoid malignancy may be more reasonable, especially when signs and symptoms discussed above persist after a minimal empiric course of antibiotics or watchful waiting. In cases with a high suspicion for lymphoma, early biopsy may be preferable. Whenever feasible, excision node biopsy is favored over needle procedures which may not preserve the architectural features useful in diagnostic evaluation. Often the affected lymph nodes will not be markedly enlarged, and identification of the biopsy target most likely to yield a diagnosis of lymphoma can be subjective. If the initially biopsied node shows only hyperplasia, further efforts to search for lymphoma should be considered, and this may necessitate an invasive surgical procedure. Occasionally, disease may be confined to the bone marrow and bone marrow biopsy should be considered when suspicion is high and lymph node biopsy alone is unrevealing.
Once the diagnosis of lymphoma has been made, it is important to adequately define clinically relevant immunophenotypic markers, such as CD20, and to complete a lymphoma staging workup. Because 17% or more of peripheral lymphoma cases in AIDS also involve the central nervous system, staging for all ARL should include brain magnetic resonance imaging (MRI) and lumbar puncture for cytological assessment of the cerebrospinal fluid (CSF).86,87 If MRI is not readily available, contrast computed tomography (CT) can be used to rule out mass lesions, but has the disadvantage of potentially missing small lesions within the brain. Also, MRI can identify leptomeningeal involvement, though sensitivity may be low.88 Flow cytometry studies on the CSF can identify occult leptomeningeal lymphoma, and where available may be useful toward ruling out this sanctuary site for lymphoma.89 Additional studies to complete lymphoma staging include CT scans of the chest, abdomen, and pelvis and bone marrow biopsy (Table 60-4). Additional studies prior to treatment initiation include complete blood count and chemistries to assess LDH, renal and hepatic function. Routine cardiac assessment should include an ECG, and if the patient has received prior anthracycline-based chemotherapy, the cardiac ejection fraction should be measured with an echo or multiple gated acquisition (MUGA) scan. CD4+ cell enumeration should be obtained at lymphoma diagnosis, since it is of prognostic importance. Fluorodeoxyglucose-positron emission tomography (FDG-PET) scans should be obtained if possible prior to therapy to identify sites of lymphoma. This can be particularly useful in assessing response in the case of residual masses seen on restaging CT scans, and early FDG-PET normalization has prognostic as well as therapeutic implications.90,91
Table 60-4 Ann Arbor Staging System
| Stage | Description |
|---|---|
| I | Single node region or lymphoid structure or single extranodal organ or site |
| IE | |
| II | Two or more nodes on the same side of the diaphragm or single nodal site with extranodal extension |
| IIE | |
| III | Nodal regions or lymphoid structure on both sides of the diaphragm |
| IIIE | Or localized involvement of an extralymphatic organ |
| IIIS | Or spleen |
| IIISE | Or both |
| IV | Bone marrow or liver or extranodal sites beyond those designated as ‘E’ |
Designate absence of constitutional symptoms with A; presence of symptoms: B (fever, sweats, unexplained weight loss greater than 10% of body weight).
From Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 31:1860–1, 1971.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


