Currently available antithrombotic drugs include (a) antiplatelet agents, which inhibit platelet activation or aggregation, (b) anticoagulants, which attenuate fibrin formation, and (c) fibrinolytic agents, which degrade the fibrin component of thrombi by catalyzing the generation of plasmin (FIGURE 110.1). Because arterial thrombi consist of platelet aggregates held together by small amounts of fibrin,1 strategies to prevent or treat arterial thrombosis focus mainly on drugs that block platelet function, but may include anticoagulants to prevent fibrin deposition. Anticoagulants are the treatment of choice for prevention of cardioembolic events in patients with atrial fibrillation or mechanical heart valves. Anticoagulants also are the drugs of choice for their prevention and treatment of venous thromboembolism (VTE) because venous thrombi are composed mainly of fibrin.1
Fibrinolytic therapy is used in selected patients with arterial or venous thrombosis. When percutaneous intervention is not feasible, systemic fibrinolytic therapy is often administered to patients with acute myocardial infarction (MI) or selected patients with ischemic stroke to rapidly restore antegrade flow in occluded arteries. Patients with massive pulmonary embolism (PE) may also benefit from systemic or catheter-directed fibrinolytic therapy. Although systemic fibrinolytic therapy is ineffective for treatment of patients with extensive deep vein thrombosis (DVT) involving the iliac and/or femoral veins, catheter-directed fibrinolysis, often in conjunction with mechanical methods of thrombus disruption, may be of benefit in some patients.2
Focusing on new antithrombotic agents, this chapter (a) identifies the limitations of current antithrombotic drugs, (b) describes the new antiplatelet and anticoagulant drugs in more advanced stages of development, focusing on their mechanism of action and the results of clinical trials, and (c) provides clinical perspective on the unmet needs in antithrombotic therapy for prevention and treatment of venous and arterial thrombosis. Fibrinolytic drugs are discussed in Chapter 112.
LIMITATIONS OF CURRENT ANTITHROMBOTIC DRUGS
New antithrombotic drugs have been developed to overcome the limitations of existing agents. Most of the advances have been in the area of antiplatelet drugs and anticoagulants. The development of new fibrinolytic agents has lagged behind.
Currently available oral antiplatelet agents include aspirin, clopidogrel, prasugrel, and ticagrelor. The efficacy of aspirin and clopidogrel has established cyclooxygenase-1 (COX-1), a key enzyme in thromboxane A2 synthesis, and P2Y12, the major ADP receptor on platelets, as important targets for antiplatelet drugs. On its own, clopidogrel has been shown to be only marginally more effective than aspirin.3 The combination of aspirin plus clopidogrel is superior to aspirin alone in patients at high risk for cardiovascular events,4, 5, 6, 7 but combination therapy is associated with a significant risk of bleeding and has yielded disappointing results in patients with stable cardiovascular disease.8, 9 Although the combination of aspirin plus dipyridamole is superior to aspirin alone for secondary prevention in patients with cerebrovascular disease,10 the efficacy of this combination is similar to that of clopidogrel.11
The limitations of existing antiplatelet drugs reflect, at least in part, their capacity to attenuate only a single pathway of platelet activation. Because platelets can be activated via multiple pathways, the potential for bypassing the inhibitory effects of these drugs remains high when there is a potent stimulus for platelet activation. Consequently, it is not surprising that breakthrough cardiovascular events occur, and these should not necessarily be labeled as simple treatment failures.
Another factor that may contribute to breakthrough cardiovascular events is individual variability in the response to antiplatelet drugs. Such variability may reflect poor compliance, pharmacogenetic factors, increased platelet turnover, drug interactions, baseline and residual platelet hyper-reactivity, as well as other factors.12, 13 Decreased responsiveness to aspirin and/or clopidogrel is common in patients with acute coronary syndrome (ACS), particularly those with diabetes.14 In patients undergoing percutaneous coronary intervention (PCI), a reduced biologic response to aspirin plus clopidogrel has been associated with a poorer outcome. The decreased response may reflect, at least in part, limited metabolic activation of clopidogrel13 in patients with reduced function variants of the CYP2C19 gene (see Chapter 111).
Although prasugrel also requires biotransformation to active metabolites, variants of the CYP2C19 gene do not appear to influence outcomes in prasugrel-treated patients.15, 16 These findings have prompted some experts to recommend tailored antiplatelet therapy based on periprocedural platelet function or genetic testing.17, 18 However, the value of this approach has not been established, and, in one study, doubling the clopidogrel dose in patients with increased platelet reactivity did not improve outcome.19 Instead, the limitations of existing antiplatelet agents has prompted the development of newer and more potent drugs, some directed against proven targets involved in platelet activation and others against new targets. These agents include novel inhibitors of the thromboxane A2 receptor, new P2Y12 antagonists, and inhibitors of protease activated receptor-1 (PAR-1), the major thrombin receptor on platelets.
On the anticoagulant front, attention has focused on the development of new oral agents to replace vitamin K antagonists (VKA).20 Rivaroxaban, a direct factor Xa inhibitor, and dabigatran etexilate, a direct thrombin inhibitor, have been licensed in many countries for short-term thromboprophylaxis after elective hip or knee arthroplasty, and dabigatran etexilate and rivaroxaban have been licensed in the United States, Canada, and Europe for stroke prevention in patients with atrial fibrillation. The pharmacologic properties of these agents are compared in Table 110.1.
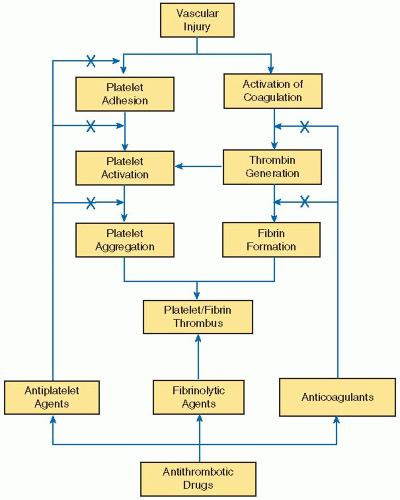
FIGURE 110.1 Targets of new antithrombotic drugs. Thrombosis occurs at sites of vascular injury by coordinated processes that result in platelet activation and activation of the coagulation system. Formation of the platelet plug represents the first step in hemostasis. Subsequent thrombin generation amplifies the platelet response because thrombin is a potent platelet agonist. Thrombin also converts fibrinogen to fibrin, and the fibrin strands stabilize the platelet aggregates, thereby forming a platelet/fibrin thrombus. Given the importance of platelets and fibrin in thrombogenesis, new antithrombotic agents target these components. New antiplatelet strategies focus on blocking platelet adhesion, activation, or aggregation. Novel anticoagulants, which target various coagulation enzymes, are geared at inhibiting thrombin generation or thrombin activity. Finally, fibrinolytic agents digest the fibrin component of the thrombus, thereby causing thrombus dissolution.
PLATELET INHIBITORS
Inhibitors of Platelet Adhesion
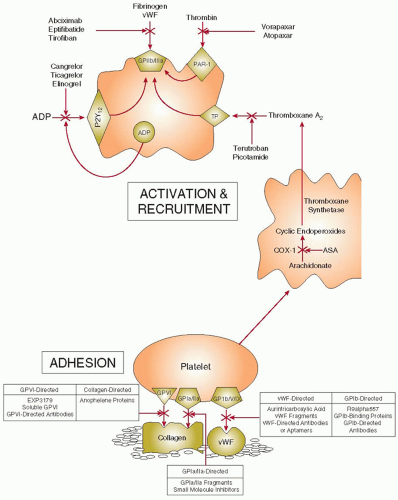
The interaction of platelets with subendothelial adhesive proteins exposed at sites of vascular injury promotes platelet signaling pathways that trigger platelet activation and adhesion. Consequently, strategies aimed at blocking platelet adhesion represent a novel antithrombotic approach. The targets for these inhibitors include von Willebrand factor (vWF), glycoprotein (GP) Ib/V/IX, GPVI, GPIa/IIa, collagen, or intracellular signaling molecules that are triggered by platelet adhesion (FIGURE 110.2).
vWF Inhibitors
Because vWF serves as the molecular glue that tethers platelets to the damaged vessel wall, vWF is an attractive target for agents that block platelet adhesion. Strategies to target vWF include (a) aurintricarboxylic acid, a heterogeneous mixture of polycarboxylic and polyaromatic polymers that bind vWF and prevent its interaction with GPIb,21, 22 (b) genetically engineered vWF fragments encompassing the GPIb-binding site, which compete with intact vWF for receptor binding,23 and (c) monoclonal antibodies or oligonucleotide aptamers directed against the A1 domain of vWF, which block the interaction of vWF with GPIb.24, 25, 26, 27 ARC1779, a DNA aptamer consisting of 40 nucleotides directed against the A1 domain of vWF, is the only one of these agents to be evaluated in humans. Polyethylene glycol was attached to the aptamer to extend its half-life. ARC1779 was well tolerated in a phase I study in healthy volunteers and has shown promise in the treatment of patients with thrombotic thrombocytopenic purpura.27
GPIb/V/IX Inhibitors
The interaction of vWF with GPIb/V/IX is an early and pivotal step in platelet adhesion. Thus, thrombus formation is abolished in GPIb-knockout mice.28 In contrast, when vWF is knocked out, thrombus formation is only modestly attenuated, presumably because other adhesive ligands substitute for vWF and mediate platelet adhesion. Strategies to target GPIb/V/IX include (a) monoclonal antibodies directed against GPIb, (b) GPIb-binding proteins isolated from the venom of Bothrops jararaca or Crotalus atrox, which compete with vWF for GPIb binding, and (c) R9alpha557, a palmitylated, cell-permeable peptide corresponding to the 557 to 569 segment of the cytoplasmic tail of GPIb that attenuates receptor signaling.29 None of these strategies has been evaluated in humans.
Table 110.1 Comparison of the pharmacologic properties of the new oral anticoagulants that are licensed or in the most advanced stages of clinical development
a One-half of renally cleared rivaroxaban is excreted as unchanged drug and one-half as inactive metabolites.
b Potent inhibitors of P-gp include quinidine, amiodarone, and verapamil. Potent inhibitors of both P-gp and CYP3A4 include azole antifungals (e.g., ketoconazole), protease inhibitors (e.g., ritonavir), and macrolide antibodies (e.g., clarithromycin). Potent CYP3A4 inhibitors include azole antifungals, macrolide antibiotics, and protease inhibitors. Potent inducer of both P-gp and CYP3A4 is rifampicin.
c Neither of these antidotes has been tested in humans. bid, twice daily; od, once daily; CYP, cytochrome P450; P-gp, P-glycoprotein efflux transporter.
GPVI Inhibitors
As a collagen receptor, GPVI plays an important part in the adhesion of platelets at sites of vascular damage. Mice deficient in GPVI exhibit reduced thrombus formation in response to ferric chloride-induced arterial injury, which exposes collagen.30 In contrast, thrombus formation in response to laser injury, which does not expose collagen, is minimally affected.30 Strategies to inhibit GPVI include (a) monoclonal antibodies directed against the receptor,31, 32 (b) soluble GPVI or a chimeric molecule consisting of GPVI linked to the Fc domain of human immunoglobulin,33, 34, 35 and (d) EXP3179, a losartan metabolite that blocks collagen-induced platelet aggregation and platelet adhesion at sites of arterial injury.36 None of these strategies has been evaluated in humans.
GPIa/IIa Inhibitors
Like GPVI, GPIa/IIa also binds collagen, thereby facilitating platelet adhesion.37 Although platelets from mice deficient in either of the GPIa/IIa subunits exhibit reduced aggregation in response to collagen, the mice have a normal bleeding time and thrombus formation in response to injury is minimally impaired.38, 39 Strategies to inhibit GPIa/IIa include (a) recombinant protein analogues of the collagen-binding site on the receptor40 and (b) small molecule receptor inhibitors.41 Neither strategy has been tested in humans.
Collagen Inhibitors
Anopheline antiplatelet protein, a protein isolated from the saliva of the Anopheles stephensi mosquito, binds collagen and inhibits its interaction with GPVI and GPIa/IIa.42 Additional work is needed to determine the therapeutic potential of this protein or the usefulness of similar antiplatelet strategies.
Inhibitors of Intracellular Signaling
An alternative strategy to targeting platelet receptors or their ligands is to block the downstream signaling pathways that trigger platelet activation. Type Ia phosphoinositide 3-kinase (PI3K) is an intracellular signaling molecule whose activation in platelets is linked to GPIIb/IIIa under high shear conditions. TGX-221, an inhibitor of the β isoform of PI3K, decreases sheardependent platelet adhesion in vitro and in animal models of arterial injury.43, 44, 45 TGX-221 has not been tested in humans.
Inhibitors of Platelet Recruitment
Strategies to block platelet recruitment have targeted the thromboxane A2 receptor, to which thromboxane A2 and its endoperoxide precursors bind, P2Y12, the most important ADP receptor, or PAR-1, the major thrombin receptor on platelets.
Thromboxane A2 Receptor Antagonists
The thromboxane A2 receptor is a G-protein-coupled receptor on platelets that is not only activated by thromboxane A2, but also by its cyclic endoperoxide precursors. Thromboxane A2 receptor antagonists were developed, at least in part, to overcome the variable response to aspirin.46 Although aspirin blocks thromboxane A2 synthesis in most individuals, elevated urinary levels of its stable metabolite, thromboxane B2, have been associated with an increased risk of cardiovascular events.47, 48 Elevated levels of urinary thromboxane B2 may reflect incomplete blockade of COX-1 in platelets and/or the shuttling of endoperoxide intermediates to platelets from other cells. The thromboxane A2 receptor antagonists include terutroban and picotamide.
FIGURE 110.2 Sites of action of new antiplatelet drugs. Platelets adhere to subendothelial collagen via glycoprotein (GP) VI and GPIa/IIa, and to von Willebrand factor (vWF) via GPIb/V/IX, respectively (top panel). Adherent platelets become activated and release thromboxane A2 and adenosine diphosphate (ADP), substances that activate adjacent platelets. By irreversibly inhibiting cyclooxygenase-1 (COX-1), aspirin blocks thromboxane A2 synthesis. Thromboxane A2 receptor (TP) antagonists attenuate platelet recruitment by preventing thromboxane A2-mediated platelet activation. ADP induces platelet recruitment by binding to P2Y12, the major ADP receptor on platelets, and triggering platelet activation. By binding to P2Y12, cangrelor, ticagrelor, and elinogrel block ADP-induced platelet activation. Thrombin, the most potent platelet agonist, binds to, cleaves, and activates protease-activated receptor-1 (PAR-1) on the platelet surface. Vorapaxar and atopaxar inhibit PAR-1 activation, thereby blocking thrombin-induced platelet activation.
Terutroban
A selective inhibitor of the thromboxane A2 receptor on platelets, terutroban (previously known as S18886), is orally active.49 Peak plasma concentrations are achieved within 1 to 2 hours of oral administration, and the drug has a half-life of 6 to 10 hours. Terutroban inhibits thromboxane A2-induced platelet aggregation in a dose-dependent fashion with maximum inhibition obtained with drug levels above 10 ng/mL—a concentration that can be achieved with daily doses of 10 to 30 mg.50
In a phase II study, peripheral arterial disease (PAD) patients were randomized to terutroban, aspirin, or placebo. Terutroban produced dose-dependent inhibition of platelet aggregation in response to thromboxane, ADP, or collagen and was at least as potent as aspirin.51 The phase III double-blind PERFORM trial compared terutroban (30 mg/day) with aspirin (100 mg/day) for secondary prevention in 19,119 patients with a recent history of ischemic stroke or transient ischemic attacks.52, 53, 54 The trial was stopped early because the rate of the primary efficacy endpoint, a composite of fatal or nonfatal ischemic stroke, fatal or nonfatal MI, or other vascular death, was 11% in both groups, and there was a slight increase in the rate of minor bleeding with terutroban compared with aspirin (12% and 11%, respectively).
Picotamide
A derivative of methoxy-isophtalic acid, picotamide not only inhibits the thromboxane A2 receptor, but also inhibits thromboxane synthetase at equivalent concentrations.55 In contrast to aspirin, picotamide does not interfere with prostacyclin production. In a double-blind, placebo-controlled study in 2,304 patients with PAD, treatment with picotamide (300 mg bid) or placebo was administered for 18 months.56 Endpoints of the study included major events (cardiovascular death, MI, stroke, or amputation) and minor events (unstable angina, transient ischemic attacks, hypertension, renal failure, or worsening of PAD symptoms). Although the intentionto-treat analysis revealed an 18.9% reduction in major plus minor events with picotamide, this difference was not statistically significant. However, the on-treatment analysis showed a 22.8% reduction in the same endpoints. Bleeding side effects were similar in the two groups. A post hoc subgroup analysis of the data from the 438 diabetic patients included in the study revealed a 45.2% reduction in major and minor endpoints with picotamide compared with placebo.57 These findings prompted a randomized trial comparing picotamide (600 mg bid) with aspirin (320 mg/day) in 1,209 diabetic patients with PAD.58 The primary efficacy endpoint was overall mortality, while the secondary endpoint was the composite of death and cardiovascular events. At 2 years, the overall mortality rates with picotamide and aspirin were 3.0% and 5.5%, respectively (relative risk [RR] 0.55, 95% confidence interval [CI] 0.31 to 0.98). Cardiovascular events occurred in 7.1% of patients given picotamide and 8.7% of those treated with aspirin. The difference in the combined endpoint of mortality plus cardiovascular events between the two groups did not reach statistical significance. Bleeding events were infrequent with both picotamide and aspirin (1.3% and 2.0%, respectively). Although these results are promising, additional studies are needed to establish the role of picotamide in diabetics with PAD.
P2Y12 Antagonists
ADP receptors on the platelet surface include P2Y12, P2Y1, and P2X1. The most important of these is P2Y12, which is the target of the thienopyridines: ticlopidine, clopidogrel, and prasugrel. However, P2Y1 also contributes to ADP-induced platelet activation, and maximal ADP-induced platelet activation requires activation of both receptors. The third receptor, P2X1, serves as an adenosine triphosphate (ATP)-gated calcium channel.
Building on the proven benefit of the thienopyridines, P2Y12 is the target for several new antiplatelet agents. In contrast to the thienopyridines, these new drugs do not require metabolic activation. Instead, they are active compounds that bind directly and reversibly to P2Y12. With no requirement for metabolic activation, the direct P2Y12 antagonists have a more rapid onset of action and produce a more potent and predictable level of receptor blockade that is unaffected by the genetic polymorphisms that influence thienopyridine absorption and/or metabolic activation. In addition, because their interaction with P2Y12 is reversible, direct P2Y12 antagonists have a more rapid offset of action than the thienopyridines, which bind P2Y12 in an irreversible fashion. The agents in most advanced stages of development include cangrelor, ticagrelor, and elinogrel (Table 110.2).
Cangrelor
An ATP analogue, cangrelor is a direct competitive inhibitor of P2Y12.58 The drug is only active when administered intravenously, and it produces almost immediate and dose-proportional inhibition of ADP-induced platelet aggregation. Cangrelor is rapidly inactivated by dephosphorylation and has a half-life of 3 to 5 minutes. Upon cessation of therapy, therefore, there is recovery of platelet function within 60 minutes.58, 59
The interaction of cangrelor with P2Y12 prevents the binding of the active metabolites of clopidogrel or prasugrel. This phenomenon complicates the transitioning of patients from cangrelor to clopidogrel or other thienopyridines, which can only exert their inhibitory effects once cangrelor dissociates from P2Y12.
Cangrelor has been evaluated in a two-part phase II trial in patients undergoing PCI.60 For part one, patients were randomized to an 18- to 24-hour infusion of cangrelor or placebo in addition to aspirin plus heparin. In the second part, patients were randomized to intravenous cangrelor or abciximab prior to PCI. In both parts, the primary endpoint, the combination of major and minor bleeding, was not different between cangrelor and the control groups (placebo or abciximab). The 30-day composite of adverse cardiac events (death, MI, or unplanned repeat coronary intervention) was also not significantly different in the two groups.
Building on these phase II data, cangrelor was investigated in two double-blind phase III trials. In the CHAMPION-PCI trial, 8,877 patients scheduled for PCI were randomized to receive intravenous cangrelor (30 µg/kg bolus followed by an infusion of 4 µg/kg/min) or placebo starting 30 minutes prior to PCI and continuing for at least 2 hours after completion of the procedure.61 All patients received aspirin. The primary efficacy endpoint, a composite of all-cause mortality, MI, or ischemia-driven revascularization within 48 hours of PCI, occurred in 7.5% of those randomized to cangrelor and 7.1% of those given placebo (hazard ratio [HR] 1.05, 95% CI 0.88 to 1.24, P = 0.59). Rates of major bleeding were similar with cangrelor or clopidogrel (3.6% and 2.9%, respectively; P = 0.06), but rates of minor bleeding were higher with cangrelor (17.6% and 15.2%, respectively; P = 0.003).
Table 110.2 Pharmacologic characteristics of direct-acting reversible P2Y12 inhibitors
Characteristic
Cangrelor
Ticagrelor
Elinogrel
Molecular weight
776
523
562
Route of administration
Intravenous
Oral
Intravenous or oral
Site of action
ADP-binding site
Site distinct from ADP-binding site
ADP-binding site
Type of inhibition
Competitive
Noncompetitive
Competitive
Time to peak activity
30 min
2 h
20 min and 12 h for intravenous and oral formulation, respectively
Frequency of oral administration
Inactive orally
Twice daily
Twice daily
Half-life
3-5 min
6-12 h
Not reported
Metabolism
Dephosphorylation
O-deethylation and oxidation
None
Elimination
Extrarenal
30% renal; 70% feces
40% renal; 60% feces
Time to recovery of platelet function
60 min
3-5 d
Not reported
Stage of development
Phase III
Completed phase III; approved in United States, Europe and Canada
Phase II
In the CHAMPION PLATFORM trial, 5,362 patients with non-ST-elevation MI or unstable angina with at least one coronary lesion amenable to PCI were randomized to receive cangrelor (in the same regimen used in the CHAMPION-PCI trial) or placebo at the time of PCI.62 The primary efficacy endpoint, which was the same as that used in CHAMPION-PCI, occurred in 7.0% of those given cangrelor and in 8.0% of patients randomized to placebo (HR 0.87, 95% CI 0.71 to 1.07, P = 0.17). Major bleeding was more frequent with cangrelor than with placebo (5.5% and 3.5%, respectively; P < 0.001). Based on the negative results of these two trials, more work is needed to determine the role of cangrelor in ACS patients undergoing PCI.
Ticagrelor
An orally active agent belonging to the cyclopentyl-triazolopyrimidine class, ticagrelor acts as a direct inhibitor of P2Y12.63 Ticagrelor binds to the receptor at a location distinct from the ADP-binding site and blocks ADP-mediated receptor activation in a noncompetitive fashion, likely through an allosteric mechanism. Because it does not require metabolic activation, ticagrelor has a rapid onset of action and achieves a level of inhibition of ADP-induced platelet aggregation within 30 minutes that exceeds that obtained with a 300 or 600-mg loading dose of clopidogrel.64 The peak inhibitory effect of ticagrelor is seen about 2 hours after a loading dose of ticagrelor of 180 mg or a maintenance dose of 90 mg twice daily.
When compared with clopidogrel in 200 aspirin-treated patients with atherosclerosis, ticagrelor, at doses of 100 or 200 mg twice daily, or 400 mg once daily, produced more rapid and more potent inhibition of ADP-induced platelet aggregation.65 The DISPERSE 2 study compared ticagrelor (90 or 180 mg twice daily) with clopidogrel (75 mg once daily) in 990 patients with non-ST-segment elevation ACS.66 All patients received aspirin. The primary endpoint, a combination of major and minor bleeding, occurred in 10.2% of patients given either dose of ticagrelor and in 9.2% of those treated with clopidogrel.
In the phase III PLATO trial, ticagrelor (180-mg loading dose followed by 90 mg twice daily) was compared with clopidogrel in 18,624 ACS patients.67 At 12 months, the primary efficacy endpoint—a composite of cardiovascular death, MI, or stroke—occurred in 9.8% of patients treated with ticagrelor and in 11.7% of those given clopidogrel (HR 0.84, 95% CI 0.77 to 0.92, P < 0.001). The rates of the individual efficacy components and of bleeding complications are summarized in Table 110.3.
An invasive strategy was planned for 72% of the 18,624 patients entered in the PLATO trial. In this subset, the primary composite endpoint occurred in 9.0% of patients randomized to ticagrelor and in 10.7% of those given clopidogrel (HR 0.84, 95% CI 0.75 to 0.94, P = 0.0025). Rates of major bleeding were similar with ticagrelor and clopidogrel (11.6% and 16.5%, respectively; P = 0.88).
PLATO-STEMI focused on the 7,544 patients with ST-elevation MI who were scheduled to undergo primary PCI.68 Of these, 75% received a stent. In this subset, definite stent thrombosis occurred in 1.6% of patients taking ticagrelor and in 2.4% of those given clopidogrel (HR 0.60, 95% CI 0.45 to 0.95, P = 0.03). Other endpoints are summarized in Table 110.3. Outcomes in the 1,899 patients in the PLATO trial who underwent coronary artery bypass grafting (CABG) surgery postrandomization were reported in PLATO-CABG.69 The rates of cardiovascular death and of all-cause mortality were reduced with ticagrelor (Table 110.3), but there was no difference in MI or stroke and no reduction in CABG-related bleeding.
Table 110.3 Summary of the results of the PLATO trial comparing ticagrelor with clopidogrel
Endpoint
Ticagrelor (180 mg Then 90 mg Twice Daily)
Clopidogrel (300-600 mg Then 75 mg Daily)
P Value
Primary endpoint (cardiovascular mortality, MI, or stroke)
PLATO
9.8% (HR 0.84, 95% CI 0.77-0.92)
11.7%
<0.001
PLATO-STEMI
9.4% (HR 0.84, 95% CI 0.75-1.01)
10.8%
0.07
PLATO-CABG
10.6% (HR 0.84, 95% CI 0.60-1.16)
13.1%
0.29
MI
PLATO
5.8%
6.9%
0.005
PLATO-STEMI
4.7%
5.8%
0.03
Cardiovascular mortality
PLATO
4.0%
5.1%
0.001
PLATO-STEMI
4.1%
9.7%
<0.01
PLATO-CABG
4.1%
7.9%
<0.01
All-cause mortality
PLATO
4.5%
5.9%
<0.001
PLATO-STEMI
5.0%
6.1%
0.05
PLATO-CABG
4.7%
9.7%
<0.01
Stroke
PLATO
1.5%
1.3%
0.22
PLATO-STEMI
Major bleeding
PLATO
11.6%
11.2%
0.43
PLATO-STEMI
9.0%
9.2%
0.76
Major bleeding not associated with CABG
PLATO
4.5%
3.8%
0.03
Fatal intracranial bleeding
PLATO
0.1%
0.01%
0.02
Overall, PLATO enrolled 18,625 patients with acute coronary syndromes. PLATO-STEMI included those 7,544 patients with ST-elevation MI who were scheduled to undergo primary PCI. PLATO-CABG included those 1,889 patients who underwent coronary artery bypass graft surgery postrandomization.
Side effects of ticagrelor include dyspnea, which is usually mild and dose related,70 asymptomatic bradycardia with ventricular pauses,67 and a modest increase in the levels of uric acid. The mechanisms responsible for these side effects are unclear. One possible explanation relates to the capacity of ticagrelor to inhibit adenosine reuptake by erythrocytes, thereby increasing circulating levels of adenosine. In addition to explaining the dyspnea and the bradycardia, the resultant adenosine-induced vasodilatation and increased myocardial perfusion could also endow ticagrelor with beneficial effects that are independent of P2Y12 blockade.
Elinogrel
A reversible P2Y12 inhibitor available in both intravenous and oral formulations, elinogrel is a competitive inhibitor of P2Y12 that blocks ADP binding to the receptor.71 Consequently, the drug inhibits platelet aggregation in response to low concentrations of ADP, but its inhibitory effects can be overcome with higher ADP concentrations. This phenomenon may endow elinogrel with a more favorable risk-benefit profile if ADP concentrations are higher with hemostatic plug formation than with intravascular thrombosis.
In the phase II INNOVATE-PCI study, 652 patients scheduled for nonurgent PCI were randomized to clopidogrel (300 to 600 mg load followed by 75 mg daily) or elinogrel (80 mg intravenous bolus followed by oral dosing of 50, 100, or 150 mg twice daily).72 The Data Safety Monitoring Board recommended discontinuation of the 50 mg oral dose and suggested increasing the intravenous dose to 120 mg. The study was not powered for efficacy, but patients given elinogrel exhibited greater platelet inhibition than those treated with clopidogrel.
Dyspnea occurred more frequently with elinogrel than with clopidogrel. Abnormal liver function tests also were more common with elinogrel, but the abnormalities appeared to resolve over time, even if the drug was continued.72, 73 The plans for phase III evaluation of elinogrel are not available.
PAR-1 Antagonists
PAR-1 belongs to a family of G-protein-coupled receptors that are activated by proteolytic cleavage.74 Human platelets express PAR-1 and PAR-4, both of which can be activated by thrombin to induce platelet secretion and aggregation. Although activation of either receptor can cause platelet aggregation independently of the other, PAR-1 and PAR-4 act synergistically to induce platelet activation. However, the affinity of PAR-1 for thrombin is 40-fold higher than that of PAR-4. Consequently, PAR-1 is activated by relatively low concentrations of thrombin, whereas PAR-4 activation requires higher thrombin concentrations. Therefore, PAR-1 is considered to be the major thrombin receptor on human platelets.74
PAR-1 also is found on endothelial cells, smooth muscle cells, fibroblasts, and cardiac myocytes.74 Thrombin-mediated activation of PAR-1 on these cells may contribute to the proliferative and proinflammatory effects of thrombin. Therefore, it is possible that PAR-1 antagonism will not only attenuate arterial thrombosis, but may also modulate other thrombin-mediated processes, including restenosis. Two orally active PAR-1 antagonists are under investigation—vorapaxar and atopaxar (Table 110.4).
Vorapaxar
A synthetic tricyclic 3-phenylpyridine analogue of himbacine, an alkaloid isolated from the bark of Australian magnolia trees that is used in several natural products, vorapaxar is a potent and specific competitive inhibitor of PAR-1.75 The drug has excellent oral bioavailability and produces dose-dependent inhibition of thrombin- or thrombin receptor agonist peptide (TRAP)-induced platelet aggregation. Vorapaxar does not affect platelet aggregation in response to other agonists nor does it affect thrombin-mediated conversion of fibrinogen to fibrin. Vorapaxar has a half-life of 126 to 269 hours and inhibits TRAP-induced platelet aggregation for up to 4 weeks. Although the binding of vorapaxar to PAR-1 is reversible, dissociation of the drug from the receptor is slow, which may explain the long half-life. Vorapaxar is metabolized by CYP3A4.74
Table 110.4 PAR-1 antagonists: comparison of the features of vorapaxar and atopaxar
Feature
Vorapaxar
Atopaxar
Molecular weight
591
608
Onset of action (h)
2
3.5
Half-life (h)
250
23
Route of elimination
Feces
Feces
Metabolism
CYP3A4
CYP3A4
Stage of development
Phase III
Phase II
In phase I studies, vorapaxar did not appear to prolong the bleeding time when administered to healthy volunteers.76 A phase II study in 1,031 patients scheduled for coronary angiography and possible PCI randomized patients to vorapaxar (at loading doses of 10, 20, or 40 mg) or placebo in a 3:1 ratio.77 A total of 573 patients underwent PCI, all of whom received aspirin, clopidogrel, and an anticoagulant (either heparin or bivalirudin). Those randomized to vorapaxar received maintenance therapy at doses of 0.5, 1.0, or 2.5 mg once daily for 2 months. The primary outcome, a combination of thrombolysis in myocardial infarction (TIMI) major and minor bleeding, occurred in 3.3% of the patients randomized to placebo and in 2.8% of those given vorapaxar. Although underpowered for efficacy, the composite of death, major adverse coronary events, or stroke occurred in 8.6% of patients randomized to placebo and in 6.2% of those given vorapaxar.
The safety of vorapaxar was also evaluated in two small studies in Japanese patients. In the first, 117 patients undergoing PCI were randomized to vorapaxar for 60 days (either 20 or 40-mg loading dose, followed by 1 or 2.5 mg daily thereafter) or placebo in addition to standard-of-care antithrombotic therapy.78 No differences were observed in the rate of the primary safety endpoint (TIMI major and minor bleeding), but compared with placebo, vorapaxar reduced the rate of nonfatal MI (42.9% and 16.9%, respectively; P = 0.013). In the second study, which included 90 patients with a recent history of ischemic stroke,79 vorapaxar (1 or 2.5 mg daily) or placebo was administered for 60 days. All patients received aspirin. Event rates were low, and vorapaxar appeared to be safe in this setting.
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



