Chapter 62 Neurological Disease
Human immunodeficiency virus (HIV)-1 infection, and particularly its late stage of severe immunodeficiency (acquired immunodeficiency syndrome, AIDS), renders the nervous system susceptible to an array of neurological disorders. In the aggregate, these can afflict virtually every component of the nervous system, manifest therefore in a variety of clinical syndromes, and contribute importantly to both morbidity and mortality. This chapter aims to provide a general view of the approach to diagnosis of these disorders. Because several of the specific neurological diseases are discussed in detail in other chapters of this volume, individual disorders are considered here chiefly in relation to issues of differential diagnosis, with greater detail confined to some of the disorders not considered elsewhere. Several recent and older reviews also discuss the individual neurological complications in more detail and provide useful, more extensive reference sections.1–8 Discussion of specific therapies is also limited in this chapter to reduce redundancy.
The frequency of neurological complications of HIV-1 infection has changed considerably in the past 10 years in the developed world, where there has been a marked reduction related to the widespread use of highly active antiretroviral therapy (HAART) and other measures.9–17 Despite this overall decrease in incidence, the spectrum of disorders afflicting those who progress to severe immunosuppression is similar to that earlier in the epidemic, with some exceptions. This chapter maintains its principal focus on these “late-stage” HIV-1-infected patients. However, as clinics follow increasing numbers of HIV-1-infected patients who do not progress to this stage of susceptibility, the frequency of encountering more “ordinary” neurological diseases similar to those affecting HIV-1-uninfected peers will rise. For these patients, the approach to neurological diagnosis will follow that of the general neurology text, rather than the algorithms presented here. Additionally, these algorithms should be followed with the usual caution that diagnosis must be carefully individualized not only with respect to variability of presenting symptoms and signs, but also recognizing that simple formulas can hardly be applicable to a patient who may present with an unusual manifestation of a common disease or a truly rare condition. Also, these diagrams necessarily present diagnostic pathways as if they are linear, when, in fact, parallel processing of several avenues is a more common and sensible approach.
GENERAL APPROACH TO DIAGNOSIS
Figure 62-1 diagrams the first steps in this anatomic approach. The clinical history usually provides a first approximation of the neuroanatomical localization (e.g., language difficulty related to dysfunction of the dominant cerebral hemisphere or numbness of both feet suggesting polyneuropathy) and the formulation of an initial neuroanatomic hypothesis. This is then tested by the neurological examination. The combination of these two bedside components usually allows not only a tentative localization but also initial consideration of the most likely diagnoses, separating CNS and PNS diseases (and those of muscle, which we have grouped with the PNS). CNS disease can be further subdivided into those affecting the brain or spinal cord.
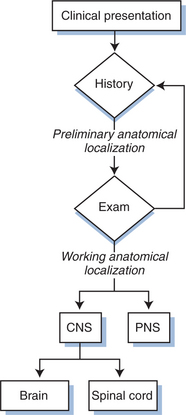
Figure 62-1 Initial approach to neurological diagnosis in HIV-1 infection. PNS, peripheral nervous system.
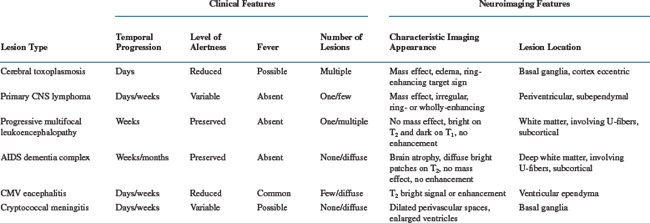
The second diagnostic element–the time course of the evolution of symptoms and signs–is also often critical to diagnosis and management. The utility of this variable has a pathobiological basis, since individual disease processes evolve over characteristic time frames, as summarized in Table 62-1. Hence, the temporal profile narrows the different possibilities. It also importantly guides the pace of diagnostic evaluation and therapeutic intervention. For example, 5 months of gradually worsening gait in a patient who walks comfortably but stiffly (as seen in the vacuolar myelopathy variant of the AIDS dementia complex) may warrant either no or only elective spinal magnetic resonance imaging (MRI). By contrast, 3 days of back pain and 4 h of leg weakness, as might accompany spinal epidural abscess, demands emergency imaging. As outlined below, the three most common causes of focal brain lesions tend to evolve at different, although overlapping, rates. This relates to the time scales of the replication of the invading organisms or of tumor cells in concert with the tempo and strength of immune responses, and can provide an initial clue to which is most likely. Exceptions to the subacute progression of most AIDS-related CNS diseases can relate to secondary developments, such as seizures, that can dramatically punctuate the course of either macroscopic or microscopic brain diseases. Hemorrhage into focal brain lesions, although far less common, can also accelerate presentation. For these reasons it is always important to define the time course of the illness and be certain that it is explained by the diagnoses being considered.
Another important contributor to “risk background” in terms of diagnosis of neurological disease in an HIV-infected patient is antiretroviral treatment status. The importance of “non-HIV-1-related” neurological diseases in treated patients with preserved immunity was emphasized earlier. Another potential implication of the larger number of treated patients with preserved immunity is that clinicians may encounter major opportunistic infections or the AIDS dementia complex at CD4+ T-lymphocyte counts higher than those reported for most such patients in earlier eras. In part this simply relates to development of this expanding population–despite continued low probabilities, the susceptible group is larger. Some differences may also relate to recovery of immune function after antiretroviral therapy. Restoration of CD4+ T-lymphocyte counts may not be followed by reconstitution of completely protective immunity against opportunistic diseases. On the other hand, effective restoration of immunity may impact on disease phenotype if vigorous host responses cause immunopathological injury. Apparent development or progression of infectious or autoimmune disorders after initiation of HAART may be due to aberrant immune responses in the setting of rapid changes in the quantity of and repertoire of circulating T cells. Such conditions seem to differ in natural history from their well-recognized counterparts occurring at extreme immunosuppression, and thus have been termed “immune reconstitution” or “restoration” diseases (IRD), or the “immune restoration inflammatory syndrome”.18–21 These disorders have proved difficult to systematically characterize, in part due to heterogeneity of clinical setting and presentation. Furthermore, based on clinical presentation alone, it may be difficult to define whether a condition relates truly to an unusual inflammatory response or simply to occurrence of a typical disease that happens to manifest soon after initiation of therapy. Such syndromes generally present within the first 3 months of HAART, mainly occur when HAART is initiated at CD4+ T-lymphocyte counts below 100 cells/μL, and have clinical features distinct from the usual natural history of opportunistic infections or autoimmune diseases in AIDS. In these conditions, addition of antiinflammatory agents such as corticosteroids may be of therapeutic benefit, though this has not yet been rigorously examined and there are no proven guidelines for management. Altered presentation of disorders involving the nervous system in the setting of immune reconstitution have recently been reported, including cryptococcal meningitis,22–24 PML,25–27 mycobacterial infections,28–31 autoimmune demyelinating neuropathies,32,33 polymyositis,34 and herpes zoster,35 among others. Although some of these disorders warrant further characterization prior to considering them entities distinct from their typical presentations, IRD in the setting of both cryptococcal meningitis and PML has been consistently observed and likely represent new manifestations of familiar disorders in the context of partially “restored” immunity, and will be discussed below. It is likely that the scope of these immune reconstitution disorders observed in the nervous system will continue to expand as more patients with advanced immunosuppression are treated with HAART.
NONFOCAL BRAIN DISEASES
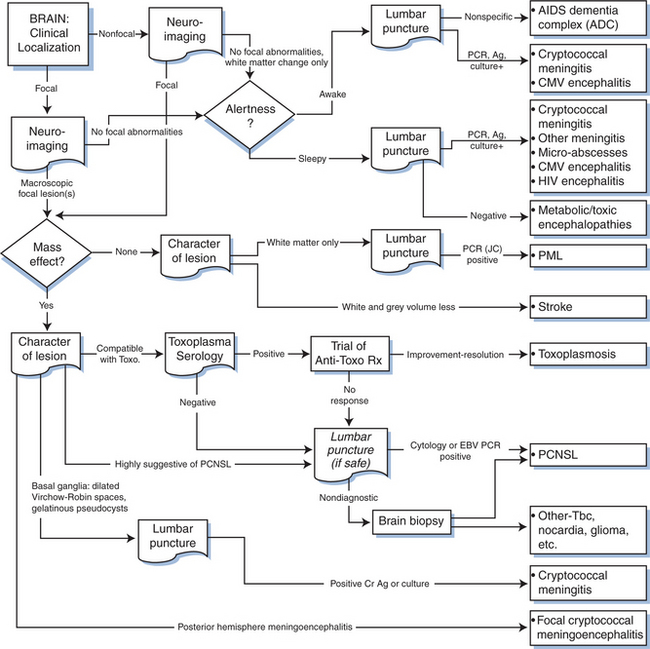
Figure 62-2 depicts a general algorithm for diagnosis of brain disease in AIDS. The upper third of this figure concentrates upon disorders that characteristically lack focal features. These conditions present with “diffuse” alterations in cognition and symmetrical motor dysfunction that is not readily explained by one or a few macroscopic focal brain lesions. There is no aphasia, apraxia, or agnosia to provide discrete cortical localization, nor hemiparesis or dysmetria to point to a lesion in a cerebral or cerebellar hemisphere. Sensory abnormalities are usually absent or reflect coincident neuropathy. These nonfocal disorders can be further segregated clinically into those in which cognition is altered in the face of preserved alertness and those in which these two elements are altered in parallel. The most important disorder in the first category is the AIDS dementia complex (ADC).
AIDS Dementia Complex
ADC is a syndrome of cognitive and motor dysfunction that has also been designated by several other synonymous terms, including HIV-associated cognitive-motor complex and the shorter terms AIDS dementia or HIV dementia.36,37 Its pediatric counterpart, which is not discussed in detail here, is most often called HIV encephalopathy.38,39 We prefer the term AIDS dementia complex for the adult form because it is simple and emphasizes that cognitive impairment is the predominating difficulty, yet also implies that this is not the only clinical manifestation and is usually accompanied by abnormal motor function and, at times, characteristic behavior abnormalities as well.40,41
ADC is believed to be caused by HIV-1 itself, rather than by another opportunistic organism, although the detailed mechanistic links between the virus and brain injury remain speculative.8,42–48 ADC is characteristically a late complication of HIV-1 infection and, particularly in its more severe form, occurs in the same context as the major clinical AIDS-defining opportunistic infections. Table 62-2 outlines the ADC staging scheme that is used to describe patients’ disease severity based on their functional capacity in cognitive and motor spheres.49,50
Table 62-2 AIDS Dementia Complex Staging
| ADC Stage | Characteristics |
|---|---|
| Stage 0 (normal) | Normal mental and motor function |
| Stage 0.5 (equivocal/subclinical) | Either minimal or equivocal symptoms of cognitive or motor dysfunction characteristic of AIDS dementia complex, or mild signs (snout response, slowed extremity movements) but without impairment of work or capacity to perform activities of daily living (ADL). Gait and strength are normal |
| Stage 1 (mild) | Unequivocal evidence (symptoms, signs, neuropsychological test performance) of functional intellectual or motor impairment characteristic of ADC but able to perform all but the more demanding aspects of work or ADL. Can walk without assistance |
| Stage 2 (moderate) | Cannot work or maintain the more demanding aspects of daily life but able to perform basic activities of self-care. Ambulatory but may require a single prop |
| Stage 3 (severe) | Major intellectual incapacity (cannot follow news or personal events, cannot sustain complex conversation, considerable slowing of all output) or motor disability (cannot walk unassisted, requiring walker or personal support, usually with slowing and clumsiness of arms as well) |
| Stage 4 (end stage) | Nearly vegetative. Intellectual and social comprehension and responses are at a rudimentary level. Nearly or absolutely mute. Paraparetic or paraplegic with double incontinence |
Adapted from Price R, Brew B: The AIDS dementia complex. J Infect Dis 158:1079, 1988; and Sidtis JJ, Price RW: Early HIV-1 infection and the AIDS dementia complex. Neurology 40:323, 1990.
On the basis of its central clinical features that include impairment of attention and concentration, slowing of mental speed and agility, concomitant slowing of motor speed and apathetic behavior, ADC has been classified among the subcortical dementias.51–54 More detailed description of its clinical features is available elsewhere.7,41,55,56 Although Figure 62-2 depicts the process of diagnosis as largely exclusionary (nonfocal neuroimaging and nonspecific lumbar puncture findings), the diagnosis should be pursued principally on the basis of its positive features, which include characteristic mental and motor findings. Additionally, although the character of the clinical abnormalities is consistent through the spectrum of its severity, it is useful to segregate the approach to diagnosis in those with milder (stage 0.5 or 1) ADC from those with more severe (stages 2–4) affliction. The major differential diagnoses often diverge in these two settings.
Mild ADC
Patients with mild ADC usually complain of difficulties with concentration and attention. They lose their train of thought in conversation or need to reread paragraphs of a book. Complex tasks previously performed “reflexively” now are more labored and take longer. Patients need to keep lists of formerly routine daily chores. These difficulties become intrusive and require compensatory strategies or changes in activities. These complaints can be similar to those of depression or of hypochondriasis. Diagnosis in these milder patients can, as a result, center on the question of whether or not there is indeed “organic” brain disease, rather than pursuit of the other nonfocal diagnoses shown in Figure 62-2. The presence of motor symptoms or signs may help support the diagnosis of ADC. These patients often have slowing of rapid finger movements (such as repeated opposing of the thumb and index tips), toe tapping and walking, along with hyperactive deep tendon reflexes and development of “release” reflexes, most notably a snout response. If there is doubt regarding either the presence or pattern of cognitive changes, formal evaluation by a neuropsychologist familiar with this condition may be helpful.57
Laboratory Diagnosis
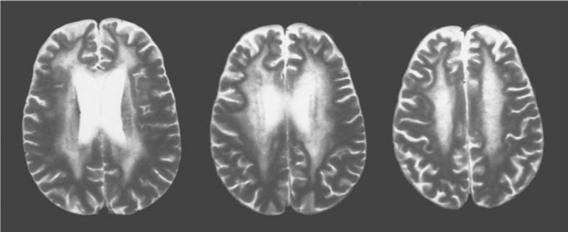
While neuroimaging is usually performed to detect evidence of alternative conditions, it may sometimes be helpful in supporting an ADC diagnosis. Most ADC patients have some degree of atrophy, detected by either computed tomography (CT) or MRI.58,59 Unfortunately, this is not diagnostically entirely specific or sensitive, since it is also found in those without clinical ADC and may be common in certain risk groups more broadly, such as substance abusers. Nonetheless, those with severe ADC usually have marked brain atrophy, with the exception of those who present with a brief, subacute course in whom atrophy may be inconspicuous or absent. Somewhat more specific are changes in white matter with increased water content noted on T2-weighted, proton density, or FLAIR sequences.60 Most common are regional changes in white matter signal, ranging from diffuse to more circumscribed “fluffy” patches of increased signal (see Fig. 62-3). If present, these tend to be more common in more severe disease, and thus are less useful in mild ADC. More recently, magnetic resonance spectroscopy (MRS) has shown abnormalities in ADC that may eventually prove useful in diagnosis and characterization of ADC.61–64 This method has not yet been tested for individual case diagnosis, but rather for clinical trials to characterize patient groups so that sensitivity and specificity for clinical application awaits further study.
CSF examination has much the same purpose as neuroimaging, primarily to screen for other disorders including cryptococcal and other meningitides, neurosyphilis, or viral encephalitis using culture, tests for antigens, and polymerase chain reaction (PCR) amplifications. It would be helpful if CSF could be used to confirm an ADC diagnosis. However, while CSF is very often abnormal, the abnormalities are not diagnostically specific. Among routine assessments, CSF protein and cell counts are frequently elevated in ADC, but these abnormalities are common in early asymptomatic systemic HIV-1 infection, especially in patients not on HAART, and may also be found in subjects with alternate diagnoses such as neurosyphilis. A number of markers of macrophage activation have been observed to be elevated in ADC, but also are not diagnostically specific. Early studies showed elevations in neopterin, beta-2-microglobulin, and quinolinic acid65–67 and more recent reports show elevations of monocyte chemoattractant protein-1 (MCP-1), other cytokines, and related inflammatory markers,68,69 but these factors are also elevated in other conditions, and their practical application to clinical diagnosis has not been carefully evaluated. In certain situations they might be useful (e.g., in diagnosis of stage 1 ADC or in distinguishing active ADC from residual injury or other static encephalopathy), but diagnostic guidelines using these tests have not been defined.
Extensive evidence supports the notion that CSF HIV-1 is compartmentalized from that in plasma, with distinct resistance genotypes and phenotypes, chemokine receptor utilization of viral quasispecies, and envelope sequences as detected by viral sequencing and cloning or heteroduplex tracking assays in the two fluids.70–72 The concept that CSF compartmentalization might reflect CNS-specific HIV-1 infection, combined with availability of quantitative HIV-1 nucleic acid amplification in CSF, has raised the question of whether viral load measurement in this compartment might help in diagnosis.73 Though there may be a correlation of CSF HIV-1 concentration with ADC severity and HIV encephalitis pathologically,74 this correlation applies only to patients with more advanced immunosuppression.75–77 In those with higher CD4+ T-lymphocyte counts, CSF viral loads may also be elevated without neurological abnormalities; additionally, CNS opportunistic infections may be accompanied by increased CSF viral load.73 Thus the CSF viral load cannot be used by itself for ADC diagnosis. There may be situations where CSF HIV-1 measurement may be useful, either for diagnosis (e.g., in the face of low CD4+ T-lymphocyte count or when the CSF viral load is higher than that of plasma) or for tracking responses to therapy. However, clear guidelines are not yet defined, and CSF viral load is not a routine part of clinical practice.
Treatment
It is clear from epidemiological studies that antiretroviral therapy can prevent ADC, and experience from limited coordinated trials and individual cases indicates that treatment can arrest or reverse the condition.7,10,15,78–88 Early studies demonstrated that zidovudine monotherapy had both therapeutic and preventative effects on ADC. Because formal clinical trials of ADC are now very difficult to implement, reports describing responses to contemporary combination antiretroviral therapy are largely anecdotal. Nonetheless, if one (1) uses the early experience with monotherapy as proof of concept, (2) extends the advantage of multidrug therapy on systemic disease to treating the CNS, (3) extrapolates from the therapeutic reduction of CSF viral load to effects on brain parenchymal infection, and (4) calls upon anecdotal (including our own personal) experience, there is a reasonable basis for considering HAART to be an effective avenue of treatment for ADC. More at issue now are refinements of therapy to better target the CNS, particularly the question of whether it is necessary that each of the components of a multidrug regimen penetrate well into the brain. Whereas several of the current nucleoside (including zidovudine, abacavir, and stavudine) and non-nucleoside (nevirapine) reverse transcriptase inhibitors penetrate relatively well (though with extracellular fluid exposures that may still be only about one-third that of plasma), most of the protease inhibitors do not.89–102 Indinavir and lopinavir are the most notable exceptions, and, in general, penetration of this class is increased by ritonavir boosting to the extent that it increases plasma concentrations. On the other hand, judging from CSF HIV responses, patients seem to respond to regimens with one or more poorly penetrating drugs, and the need for such drug penetration may vary in different types of patients. Also, drug levels in CSF do not necessarily indicate exposure and intracellular effects on the critical target cells, including particularly brain macrophages. As a result, in the absence of clearer guidelines, we recommend that drugs for symptomatic ADC patients be selected with the following general priorities: (1) assure susceptibility of virus isolates to the individual drugs just as in treatment of systemic infection, including susceptibility to CSF isolates in cases with prior drug exposure and suspected “compartmentalized” infection; (2) use an aggressive regimen, usually with four or more drugs, including nucleosides; and, finally (3) where possible, choose at least two drugs with more favorable CNS penetration including those listed above.
Additional approaches to treat ADC can be grouped under the category of adjuvant treatments. These range from various forms of symptom management (e.g., cautious neuroleptics or mood stabilizers to relieve behavioral symptoms and signs) to efforts at attenuating the disease process by interfering with various endogenous neurotoxic pathways. The latter has led to clinical trials of candidate neuroprotective agents without firm evidence of efficacy.103–105
Other Nonfocal Brain Disorders
Most nonfocal brain disorders complicating HIV-1 infection other than ADC are accompanied by concomitant depression of both alertness and cognition. Overall, the most common in this category are the toxic encephalopathies related to sedative, narcotic, and other CNS-acting medications. Metabolic encephalopathies are also common and relate to failure of systemic organs–hypoxia, renal or hepatic failure, and the like.
Among opportunistic infections, the most common “nonfocal” infection causing general confusion with a picture similar to metabolic encephalopathy is CMV encephalitis.106–113 Like other organ afflictions with this herpesvirus, CMV encephalitis is becoming rare in patients taking HAART. Additionally, its clinical spectrum has not been sharply defined. Autopsy studies earlier in the AIDS epidemic showed histological evidence of CMV infection in about one quarter of brains.114–117 However, the extent to which these lesions, some quite mild, contributed to clinical abnormalities was uncertain in many cases. On the other hand, it is also clear that some patients develop severe CMV encephalitis with distinct morbidity and mortality. Thus the key issue was not whether CMV caused symptomatic brain infection in AIDS, but rather what was the importance of the milder end of the spectrum. Also, CMV infection can coincide with ADC, and it may be difficult to discern which process is responsible for particular clinical manifestations. Clinical-pathologic studies have pointed out certain features that associate with CMV encephalitis. These include subacute onset, frank confusion or delirium, hyponatremia, and, more specifically, periventricular abnormalities on MRI (increased water signal or enhancement with contrast). Some patients have distinct focal features, including nystagmus, ataxia, and ocular motor palsies, indicating brain stem involvement; seizures are likely more common than with ADC. Unusual cases of CMV have more prominent focal features and may even have focal lesions detected on neuroimaging that exceed 1 cm in diameter.118 CSF may vary from bland fluid to pleocytosis with polymorphonuclear or mononuclear predominance and high protein. Diagnosis has been revolutionized by nucleic acid amplification techniques, and detection of CMV DNA provides a sensitive test.106,119–123 Most, but not all, patients have CMV disease in other organs, which often helps to support the diagnosis. Therapy is with one or more of the anti-CMV drugs discussed elsewhere in this volume.
Other diseases that cause widespread microscopic pathology may present a similar picture, including the encephalitic form of cerebral toxoplasmosis that presents without distinct focality and may be accompanied by a nearly normal CT scan.124,125 This is usually a fulminant infection associated with multiple microabscesses, with little inflammatory or tissue reaction. In cases of rapid-onset encephalitis of this type, empiric antitoxoplasma therapy may be justified while diagnostic evaluation is underway. Diffuse CNS dysfunction may also be caused by disseminated intravascular coagulation in the septic or otherwise gravely ill patient. Herpes simplex encephalitis in the AIDS patient may also present as a diffuse encephalopathy and differ from the characteristic focal encephalitis of the immunocompetent patient.119,126–129 Neuroimaging may be negative. PCR amplification of HSV in the CSF is likely the most sensitive diagnostic tool, but test results are often delayed; if this diagnosis is suspected, empiric therapy with intravenous acyclovir may be warranted until results are available.
Meningitis and Headache
As in other settings, meningitis in AIDS may present with confusion and altered consciousness as the predominating manifestations. In the case of pyogenic bacterial meningitis caused by Listeria monocytogenes or Streptococcus pneumoniae, which are more common complications of HIV in developing regions,130 headache and stiff neck are usually present and push the clinician to lumbar puncture. However, these findings may be less conspicuous or even absent at onset in cryptococcal meningitis, the most common meningitis in this setting. Frequently there is only a mild inflammatory reaction in the CSF compared to the burden of microorganisms recovered (see Chapter 43).131 Hence, even relatively low suspicion of the latter should trigger lumbar puncture and analysis for cryptococcal antigen, along with diagnostic studies for bacteria and other fungi, particularly when the CD4+ T-lymphocyte count is low. Recently, CNS cryptococcal disease with predominant inflammatory characteristics has been described in the setting of immune reconstitution on HAART. Within 6 weeks of starting potent HAART, some patients with previously undetected disease develop clinically apparent, rapidly progressive meningitis characterized by neck stiffness, headache, nausea, vomiting, and a prominent CSF pleocytosis.24 Unusual manifestations of cryptococcal disease have been observed in patients with a known cryptococcal diagnosis by culture or antigen positivity, started on HAART soon after antifungal treatment.22 IRD in these patients was characterized by various forms of sterile inflammation, including clinical syndromes of meningismus, headache, nausea, vomiting, increased intracranial pressure, and lack of organisms on CSF culture. Hypothesized mechanisms for these presentations include an atypical response to a previously clinically latent infection, or an enhanced (and immonopathological) response to the presence of low-grade cryptococcal antigenemia in the CSF. These syndromes present a departure from the more typical indolent course of cryptococcal meningitis, and have raised issues of when to initiate HAART in patients with known cryptococcal disease.132
CSF abnormalities such as a mild mononuclear pleocytosis (10–400 cells/μL) and elevated protein, accompanied by a variable degree of overt symptoms may be seen in patients with HIV-1 infection and neurosyphilis. Syphilis is now endemic in many metropolitan areas in the developed world133,134 and is a major co-infection for patients with HIV.135 The clinical presentations of neurosyphilis include meningeal involvement with prominent cranial neuropathies, meningovascular disease with stroke, uveitis from ocular disease, dementia from general paresis, and ataxia and neuropathic pain from tabes dorsalis.136 Of these, HIV-infected patients most commonly present with meningeal or ocular syphilis. Diagnosis of neurosyphilis is accomplished mainly by obtaining CSF in subjects found to have a positive serum rapid plasma reagin (RPR) test, examining the fluid for positive VDRL and RPR titers.137 Unfortunately, CSF VDRL is only 30–70% sensitive in cases of neurosyphilis,138,139 so more conservative algorithms use diagnostic criteria of a positive serum RPR and elevated CSF protein or white blood cells in the setting of HIV-1 infection. As we have noted, CSF abnormalities in HIV-1-infected patients may be difficult to interpret, primarily because many (mostly not on HAART) with seemingly asymptomatic HIV have pleocytosis and elevated protein, likely due to HIV itself. As a result, new, specific methods are needed to define the patient truly afflicted with neurosyphilis, in order to prioritize appropriate therapy which involves 10–14 days of IV or IM penicillin. A recent report has suggested that detection of an elevated proportion of B lymphocytes in the CSF, in combination with measurement of CSF fluorescent treponemal antibody (FTA), may be more sensitive than CSF VDRL and more specific than the presence of pleocytosis for diagnosis of neurosyphilis.140
Mycobacterium tuberculosis is a major cause of meningitis, neurological morbidity, and death in regions where tuberculosis is endemic or epidemic, though less common in the Western world.130,141,142 The typical presentation of tuberculous (TB) meningitis pursues a somewhat protracted course (days to weeks), with CSF demonstrating increased lymphocyte predominance, elevated protein levels, and low glucose. Recent studies of TB meningitis in HIV-infected patients have demonstrated that HIV-infected patients may have more extrameningeal TB, more infarcts, and fewer meningeal signs and symptoms than seen in the typical course of disease. Furthermore, CSF may demonstrate either acellularity, neutrophil predominance, or normal protein in more than 10% of AIDS patients with TB meningitis.141,142 Treatment requires evaluation for drug-resistant strains in some areas, multidrug therapy, and in some cases ventriculoperitonal shunting for hydrocephalus, though this has not been shown to be of clear overall benefit.143
Although headache is a common symptom in AIDS, many patients with this complaint do not suffer bacterial or fungal infections.144–150 As discussed above, in some individuals with HIV-1 infection lumbar puncture will detect a pleocytosis sufficient to warrant the designation of “aseptic meningitis”. However, use of this term for a relatively common laboratory finding in otherwise asymptomatic individuals can be problematic, and one should probably distinguish patients who develop meningitic symptoms and exhibit these types of CSF findings from those in whom they are incidental. In an earlier report, Hollander divided patients who presented with headache and CSF pleocytosis into two groups, those with an acute presentation and those with a more chronic course.151 Because of the high prevalence of asymptomatic pleocytosis in HIV-1 infection, the relationship between elevated CSF cells and headache is not always clear, and in some the symptom and the laboratory finding may indeed be unrelated.152 In our own studies of treatment interruption in which we have noted a resurgence of HIV-1 in CSF accompanying that in plasma, we have been struck not only by the robust lymphocytic cell response but also by the fact that this response is clinically silent.153 No formal study has addressed the question of whether headache in HIV-1 infection responds to HAART, though pleocytosis without other causes characteristically resolves after institution of HAART in parallel with clearing of CSF virus.71,154 Further complicating this issue, a clinically similar headache can manifest in HIV-1-infected patients in the absence of pleocytosis; this was common enough to have been designated “HIV headache”.155,156 This headache can have a migrainous quality with periodicity, photophobia, and nausea. Treatment is empiric and may include agents active in prophylaxis of migraine, such as anticonvulsants, tricyclics, or calcium channel blockers.
FOCAL BRAIN DISEASES
The diagnosis of macroscopic brain disorders begins with clinical recognition of symptoms and signs that indicate the presence of focal brain dysfunction. These include hemispheric (hemiparesis, hemianopsia, aphasia, and apraxia) or brain stem-cerebellar (vertigo, ataxia, diplopia, bilateral pyramidal signs, and the like) dysfunction. Symptoms and signs may be prominent or subtle, and more than one focal lesion affecting “eloquent” brain structures may confuse exact localization. The first step in evaluation is usually neuroimaging both to confirm the presence of detectable focal lesions and to further define their character (see Fig. 62-2). Only rarely will focal lesions escape detection by MRI; in such patients either the imaging is done too early to discern abnormalities (e.g., the rare patient with PML at its earliest stage), or the abnormalities are microscopic and below the limit of image detection. Some metabolic disorders may present with focal features (e.g., nonketotic, hyperglycemic hyperosmolar encephalopathy). In addition, microscopic disease, encephalitis, or meningitis can cause seizures that transiently expand the zone of physiologic dysfunction beyond that caused by the lesion itself, with residual focal deficits that are slow to clear.
Among the focal brain disorders afflicting AIDS patients, PCNSL, cerebral toxoplasmosis, and PML are most common, and most diagnostic efforts relate to distinguishing these three diseases. Since neurological symptoms and signs are determined by localization, the particular deficits are not helpful in their segregation. However, each of the three tends to have a different temporal profile (see Table 62-1). In general, toxoplasmosis evolves most rapidly, presenting within a few days after the onset of symptoms. PCNSL usually evolves somewhat more slowly, with 1 to 3 weeks separating the onset of first symptoms and presentation to the physician. PML advances even more slowly and may take several weeks to a few months before the patient seeks medical evaluation. Associated constitutional symptoms also tend to differ. Patients with toxoplasmosis are more often febrile, appear generally ill, and are lethargic or sleepy; they also more commonly complain of headache. This contrasts with both the PCNSL and PML patients, who are constitutionally asymptomatic unless they suffer an additional overt infection.
Progressive Multifocal Leukoencephalopathy
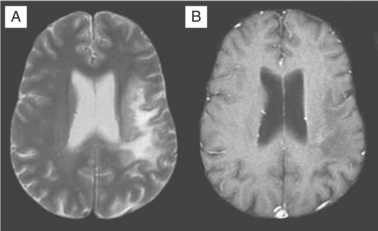
Despite these clinical differences, neuroimaging is needed to further define diagnosis of these focal disorders. MRI usually readily identifies PML.156–160 PML is caused by JC virus brain infection and is characterized by demyelination and loss of tissue rather than an expanding mass effect.161,162 Areas of demyelination that follow death of infected oligodendrocytes coalesce to leave behind areas of lost white matter and even cavitation. PML lesions usually begin as small foci that expand concentrically, either singly or in several sites. Inflammation and surrounding edema is usually absent or scant, and consequently the typical MRI of PML shows one or more lesions afflicting predominantly or exclusively the white matter in which there is increased signal (white) on T2-weighted images but diminished signal (black) on T1-weighted images (see Fig. 62-4). Lesions have a predilection to involve the white matter adjacent to the cortex but can be located anywhere. When contrast enhancement is present (likely in <10% of cases), it tends to have a delicate, lacy appearance. There are two principal differential diagnoses to consider with this type of scan result, both of which are usually easily eliminated by correlation with the clinical presentation and by observing their evolution over time. The first is ADC, which can be associated with focal white matter changes. However, ADC characteristically involves deeper white matter, lesions usually are not black on T1-weighted images, and, most importantly, localized image abnormalities are not accompanied by corresponding focal neurological deficits. The second is cerebral infarction. However, in the latter, the gray matter is also affected, the distribution of the lesion follows a vascular territory, and the clinical evolution occurs in hours rather than weeks.
A common clinical question in these patients is, “What level of diagnostic certainty is necessary for clinical management of PML?” For the typical case in which the MRI shows the abnormalities outlined above (confirmed by an experienced neuroradiologist) and these imaging findings can be correlated with the clinical picture, a clinical diagnosis of PML is highly probable and may not require further diagnostic testing. CSF examination should probably be done routinely to eliminate diagnostic surprises such as neurosyphilis or vasculitis, depending upon the individual patient’s findings. CSF also allows PCR confirmation of local JC virus infection detected in the CSF with ∼75% sensitivity but very high specificity for PML,163–168 though the sensitivity of this test may be lower in the setting of HAART.169 If other measures fail and the case is atypical, brain biopsy should be undertaken for final confirmation.
There is no proven specific treatment for PML. However, remission has been clearly documented. Early in the epidemic, Berger and colleagues noted spontaneous remission in the absence of treatment, and following a number of case reports documenting remission in PML patients treated with HAART,170 retrospective studies comparing the outcome of this opportunistic infection before and after HAART have demonstrated that about half of PML patients do well after starting HAART, with arrest of progression and an element of improvement in some.171–175 Radiographic improvement has also been documented by MRI.176 The theoretical interpretation of this response, which is in keeping with earlier reports of PML remission in non-AIDS patients, is that HAART restores the host’s capacity to mount an effective immune response to JC virus. The current standard of therapy for PML therefore begins with institution or modification of HAART, which is then assessed by the standard measures of viral load reduction and CD4+ T-lymphocyte cell increase.
For those cases that fail to respond, the remaining treatment options are experimental. Unfortunately, the history of PML includes reports of several therapies that initially showed promise but failed when examined prospectively in a larger trial.177 Most recently, interest has centered upon the use of cidofovir, an antiviral active against CMV. Studies of this drug in PML have been uncontrolled and the results are conflicting; a recent prospective multicenter trial found no benefit of cidofovir in terms of neurological examination score after 8 weeks of treatment.178–181
In the setting of immune reconstitution, new phenotypes of CNS disease referable to infection with JC virus have been described. Neurological deficits due to PML may occur around the time of immune reconstitution, either newly developing after HAART, or worsening in the context of immune restoration with increased CD4+ T-lymphocyte counts.25,182,183 On MRI scans, these lesions may appear typical of PML in location and morphology, but may also show edema, mass effect, and peripheral enhancement with gadolinium, suggesting a local immune reaction and breakdown of the blood–brain barrier and inflammation. This inflammatory nature of PML immune reconstitution disease has been confirmed by biopsies demonstrating an atypical inflammatory reaction (perivascular lymphomonocytic infiltration of CD8+ T-lymphocyte suppressor cells and CD20+ B cells) in cerebral white matter in patients with PML who had dramatic clinical worsening soon after HAART was initiated.26 Finally, a recent report describes two patients with known PML who had paradoxical fatal worsening after initiating HAART. Because of atypical presentations, fulminant progression, and negative JC virus PCR from the CSF, brain biopsies were performed, demonstrating dramatic lymphocytic infiltrates and JC virus detected in brain tissue through PCR.27 In several of these reports, antiinflammatory therapy with corticosteroids was administered as adjunctive therapy to HAART, but did not seem to alter the course of disease. The paradox of immune reconstitution disease is especially manifest in these examples of progression of PML in the setting of HAART, since otherwise the prognosis of this disease has been overall dramatically improved in the era of combined antiretroviral therapy.
A recent report has expanded the cellular tropism of JC virus beyond the oligodendrocyte to the granule cell layer of the cerebellum, suggesting that diffuse cerebellar dysfunction and atrophy in patients with AIDS may be mediated by cytolytic neuronal infection by JC virus even in the absence of demyelinating lesions.184,185
Cerebral Toxoplasmosis and PCNSL
The other two common focal brain lesions, cerebral toxoplasmosis and PCNSL, in contrast to PML, usually are defined radiologically as having mass effect (an expansion of tissue) with surrounding edema. Figure 62-2 outlines an approach to diagnosis of focal mass lesions that is aimed particularly at the early diagnosis of brain lesions other than toxoplasmosis. Because cerebral toxoplasmosis is nearly always due to reactivated Toxoplasma gondii infection, greater than 95% of such patients have detectable serum antibody to this organism.125,186–188 Hence, if the radiographic character of the lesion(s) is compatible with toxoplasmosis and the blood serology is positive, the patients are subjected to a trial of antitoxoplasma therapy. They improve clinically within days to (at most) 2 weeks, and this is followed with radiographic documentation (Fig. 62-5). Typically, neuroimaging shows contrast-enhancing lesions (usually multiple, but they can be single) involving the cortex or deep gray nuclei (basal ganglia and thalamus); the lesions often have a border of strong contrast enhancement, and there may be an “eccentric target” sign.189,190 Because prophylaxis against Pneumocystis carinii pneumonia with trimethoprim–sulfamethoxazole is also active in preventing cerebral toxoplasmosis, this variable is also considered in the probability of this diagnosis.163 Indeed, its widespread use, and more recently the use of HAART, has reduced the incidence of cerebral toxoplasmosis.13 However, it is still a prevalent disorder in many settings, chiefly in individuals who have not been receiving ongoing medical care.191
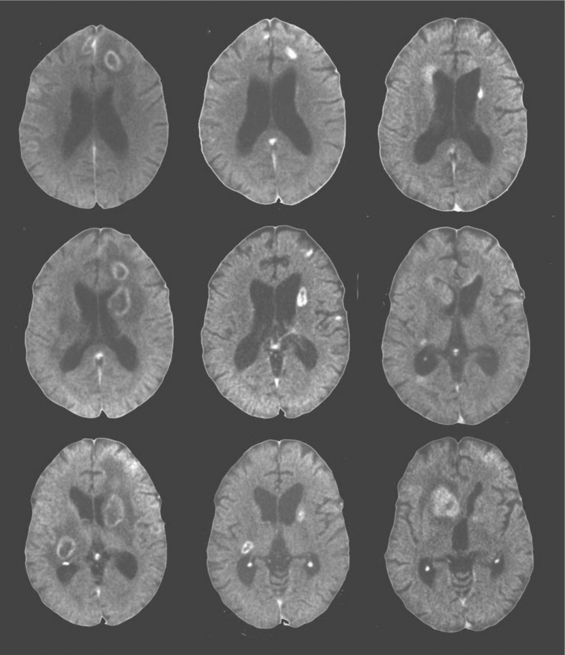
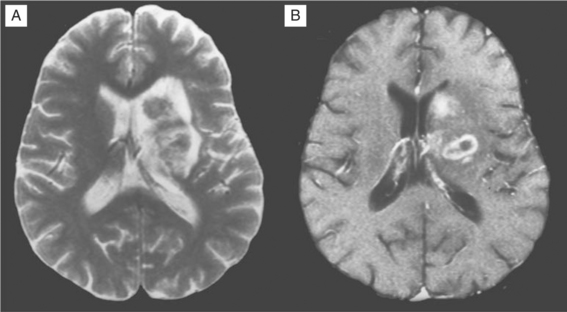
Because the outcome of PCNSL is likely influenced by how early diagnosis is made and treatment started, it is important to pursue this diagnosis aggressively. In this respect, it is not reasonable to simply undertake a trial of toxoplasmosis therapy in all AIDS patients with mass lesions, as had been recommended by some in the past. In patients with negative toxoplasma blood serology, neuroimaging abnormalities that suggest PCNSL or with radiographically and clinically atypical features, we advocate early brain biopsy, though usually after CSF examination if lumbar puncture is judged to be safe. Imaging abnormalities that favor PCNSL include deep lesions involving the white matter, including the corpus callosum; subependymal extension of lesions along the ventricular walls; and diffuse or weak contrast enhancement rather than the ring-like appearance of toxoplasmosis (Figs 62-5 and 62-6).190,192 In most settings, brain biopsy is necessary for certain diagnosis of PCNSL. PCR detection of Epstein–Barr virus (EBV) DNA sequences in CSF has been reported to enhance diagnosis, perhaps to the point of substituting for biopsy.4,163,193,194 However, the accuracy of this test through different commercial laboratories needs to be confirmed by more extended experience. Conventional cytology is much less commonly helpful.195

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


