Neuroblastoma, a neoplasm of the sympathetic nervous system, is the second most common extracranial malignant tumor of childhood and the most common solid tumor of infancy. Neuroblastoma is a heterogeneous malignancy with prognosis ranging from near uniform survival to high risk for fatal demise. Neuroblastoma serves as a paradigm for the prognostic utility of biologic and clinical data and the potential to tailor therapy for patient cohorts at low, intermediate, and high risk for recurrence. This article summarizes our understanding of neuroblastoma biology and prognostic features and discusses their impact on current and proposed risk stratification schemas, risk-based therapeutic approaches, and the development of novel therapies for patients at high risk for failure.
Neuroblastoma, a neoplasm of the sympathetic nervous system, is the second most common extracranial malignant tumor of childhood and the most common solid tumor of infancy. Neuroblastoma is a heterogeneous malignancy with prognosis ranging from near uniform survival to high risk for fatal demise. Neuroblastoma serves as a paradigm for the prognostic utility of biologic and clinical data and the potential to tailor therapy for patient cohorts at low, intermediate, and high risk for recurrence. Overall survival is excellent for patients who have low- and intermediate-risk neuroblastoma with a general trend toward minimization of therapy. In contrast, a marked intensification of therapy has led to only incremental improvement in survival for high-risk disease because less than 40% of high-risk patients survive. Chemotherapy and radiotherapy resistance remain the hallmark of failure. This article summarizes our understanding of neuroblastoma biology and prognostic features and discusses their impact on current and proposed risk stratification schemas, risk-based therapeutic approaches, and the development of novel therapies for patients at high risk for failure.
Epidemiology and cause
The incidence of neuroblastoma per year is 10.5 per million children less than 15 years of age. Neuroblastoma accounts for 8% to 10% of all childhood cancers and for approximately 15% of cancer deaths in children. There seems to be no significant geographical variation in the incidence between North America and Europe, and there are no differences between races. Neuroblastoma occurs slightly more frequently in boys than girls (ratio 1.2:1). The incidence peaks at age 0 to 4 years, with a median age of 23 months. Forty percent of patients who present with clinical symptoms at diagnosis are under 1 year of age, and less than 5% with clinical symptoms are over the age of 10 years. Cases of familial neuroblastoma have been reported. Environmental factors are implicated in the development of neuroblastoma (eg, paternal exposure to electromagnetic fields or prenatal exposure to alcohol, pesticides, or phenobarbital). A potential relationship with assisted pregnancies has also been made. None of these environmental factors has been confirmed in independent studies.
Screening for neuroblastoma was pioneered by Japanese investigators who demonstrated that asymptomatic tumors could be detected in infants by measurement of urinary catecholamine metabolites. The implementation of infant screening for neuroblastoma resulted in a doubling of neuroblastoma incidence to 20.1 per million children, and the tumors detected possessed favorable biological characteristics. Although the outcome for the children with the detected tumors was excellent, these studies were not population based and did not demonstrate a resultant reduction in neuroblastoma mortality rates. The Quebec Neuroblastoma Screening Project and the German Neuroblastoma Screening Study were designed to identify whether screening a large cohort of infants for neuroblastoma at the ages of 3 weeks, 6 months, and 12 months could reduce the population-based incidence of advanced disease and mortality. These studies demonstrate that screening for neuroblastoma at or under the age of 1 year identifies tumors with a good prognosis and molecular pathology, doubles the incidence, and fails to detect the poor-prognosis disease that presents clinically at an older age.
Genetic predisposition
Neuroblastoma can occur in patients affected with other neural crest disorders or malignancies, such as Hirschsprung disease, neurofibromatosis type 1, and congenital central hypoventilation syndrome. Genomic linkage studies have not found evidence of a link between Hirschprung disease and neuroblastoma development. The co-occurrence of neuroblastoma and von Recklinghausen disease is of interest because both disorders are deviations of normal neural-crest cell development in the embryo. An analysis of the reported coincidence of neuroblastoma and neurofibromatosis indicates that most of these cases can probably be accounted for by chance. The PHOX2B gene is the major disease gene for the congenital central hypoventilation disorder, and constitutional PHOX2B mutations have been identified in familial neuroblastoma cases and in 2.3% of patients who have sporadic neuroblastoma.
Several cases of constitutional chromosome abnormalities have been reported in individuals who have neuroblastoma, but no consistent pattern has emerged. Constitutional abnormalities involving the short arm of chromosome 1 have been reported for three neuroblastoma cases, possibly implicating chromosome 1 p loci in neuroblastoma predisposition. A report that familial neuroblastoma is not linked to 1p36 indicates that the predisposition locus for familial cases lies elsewhere.
Familial forms of neuroblastoma are rare, accounting for about 1% of all cases. There are few reported pedigrees of familial neuroblastoma. In those families, the median age at diagnosis is 9 months, as opposed to 2 to 3 years in sporadic cases. An increased incidence of multiple primary tumors is also apparent. Analysis of the pedigree structures suggests a dominant mode of inheritance with low penetrance. Germline mutations in the ALK oncogene are the main cause of familial susceptibility to neuroblastoma in otherwise healthy families. ALK is predicted to function as an oncogene in the pathogenesis of neuroblastoma. Disease-causing mutations are found almost exclusively within the tyrosine kinase domain of the ALK that lead to constitutive phosphorylation and activation of the ALK protein.
Genetic predisposition
Neuroblastoma can occur in patients affected with other neural crest disorders or malignancies, such as Hirschsprung disease, neurofibromatosis type 1, and congenital central hypoventilation syndrome. Genomic linkage studies have not found evidence of a link between Hirschprung disease and neuroblastoma development. The co-occurrence of neuroblastoma and von Recklinghausen disease is of interest because both disorders are deviations of normal neural-crest cell development in the embryo. An analysis of the reported coincidence of neuroblastoma and neurofibromatosis indicates that most of these cases can probably be accounted for by chance. The PHOX2B gene is the major disease gene for the congenital central hypoventilation disorder, and constitutional PHOX2B mutations have been identified in familial neuroblastoma cases and in 2.3% of patients who have sporadic neuroblastoma.
Several cases of constitutional chromosome abnormalities have been reported in individuals who have neuroblastoma, but no consistent pattern has emerged. Constitutional abnormalities involving the short arm of chromosome 1 have been reported for three neuroblastoma cases, possibly implicating chromosome 1 p loci in neuroblastoma predisposition. A report that familial neuroblastoma is not linked to 1p36 indicates that the predisposition locus for familial cases lies elsewhere.
Familial forms of neuroblastoma are rare, accounting for about 1% of all cases. There are few reported pedigrees of familial neuroblastoma. In those families, the median age at diagnosis is 9 months, as opposed to 2 to 3 years in sporadic cases. An increased incidence of multiple primary tumors is also apparent. Analysis of the pedigree structures suggests a dominant mode of inheritance with low penetrance. Germline mutations in the ALK oncogene are the main cause of familial susceptibility to neuroblastoma in otherwise healthy families. ALK is predicted to function as an oncogene in the pathogenesis of neuroblastoma. Disease-causing mutations are found almost exclusively within the tyrosine kinase domain of the ALK that lead to constitutive phosphorylation and activation of the ALK protein.
Pathology
Cell of Origin and Cancer Stem Cell Hypothesis
The peripheral neuroblastic tumors (pNTs), including neuroblastoma, belong to the “small blue round cell” neoplasms of childhood. They are derived from progenitor cells of the sympathetic nervous system: the sympathogonia of the sympathoadrenal lineage. After migrating from the neural crest, these pluripotent sympathogonia form the sympathetic ganglia, the chromaffin cells of the adrenal medulla, and the paraganglia, reflecting the typical localizations of neuroblastic tumors.
The mechanisms causing persistence of embryonal cells that later give rise to pNTs are mainly unknown. Defects in embryonic genes controlling neural crest development are likely to underlie the unbalanced proliferation and disturbed differentiation of neuroblastoma. These defects cause a disruption of the normal genetic differentiation program, resulting in an early or late differentiation block. The classic histopathologic pNT subtypes of neuroblastoma, ganglioneuroblastoma, and ganglioneuroma reflect a spectrum of maturation ranging from tumors with predominant undifferentiated neuroblasts to those largely consisting of fully differentiated neurons surrounded by a dense stroma of Schwann cells.
A hallmark of neuroblastoma is cellular heterogeneity. Although the presence of phenotypically diverse cells could be explained by ongoing mutagenesis, the cancer stem cell hypothesis has provided an intriguing alternative explanation for neuroblastoma heterogeneity. This hypothesis suggests that rare multipotent stem cells with indefinite potential for self-renewal drive the onset and growth of tumors. Although the existence of cancer stem cells in leukemia and some solid tumors has been established, neuroblastoma stem cells have not been clearly identified. Developmental programs controlling self-renewal in neuronal stem cells, including the Notch, Sonic hedgehog, and Wnt/β-catenin pathways, have been implicated in embryonal tumorigenesis. It is conceivable that neuroblastoma stem cells arise from normal neural crest stem cells, partly preserving and partly dysregulating these pathways. The identification and characterization of cancer stem cells in neuroblastoma should permit a targeted approach to more effective treatment.
Histopathologic Assessment
Primary, pretreatment tumor specimens obtained by open biopsy are optimal material for histologic examination and prognostic evaluation. The typical neuroblastoma is composed of small, uniformly sized cells containing dense hyperchromatic nuclei and scant cytoplasm. The Homer-Wright pseudorosette composed of neuroblasts surrounding areas of eosinophilic neuropil is seen in up to 50% of cases. Distinguishing pNTs from other “small blue round cell” tumors often requires techniques beyond hematoxylin-eosin staining and light microscopy. In the immunohistochemical diagnosis of pNTs, positive staining for neural markers, including neuron-specific enolase, synaptophysin, neurofilament protein, ganglioside GD2, chromogranin A, and tyrosine hydroxylase, combined with negative staining for markers of other small-round-cell tumors should be considered. Electron microscopy typically demonstrates dense core, membrane-bound neurosecretory granules, microfilaments, and parallel arrays of microtubules within neuritic processes (neuropil).
As early as 1963, Beckwith and Perrin suggested a natural history of pNTs that might include involution (regression) and maturation. This hypothesis was based on their observation of “in situ neuroblastoma,” an adrenal lesion of microscopic size that is cytologically identical to typical neuroblastoma and is detected in infants with a frequency of 50 times the expected incidence of primary adrenal neuroblastoma. The concept of Beckwith and Perrin has been adopted and incorporated in the International Neuroblastoma Pathology Classification (INPC). The INPC was established in 1999 by adopting the original system proposed by Shimada in 1984. The INPC was revised in 2003. The INPC distinguishes a favorable histology group from an unfavorable histology group of pNTs by applying the concept of age-dependent normal ranges of morphologic features, such as Schwannian stromal development, grade of neuroblastic differentiation, and mitosis-karyorrexis index ( Fig. 1 ).
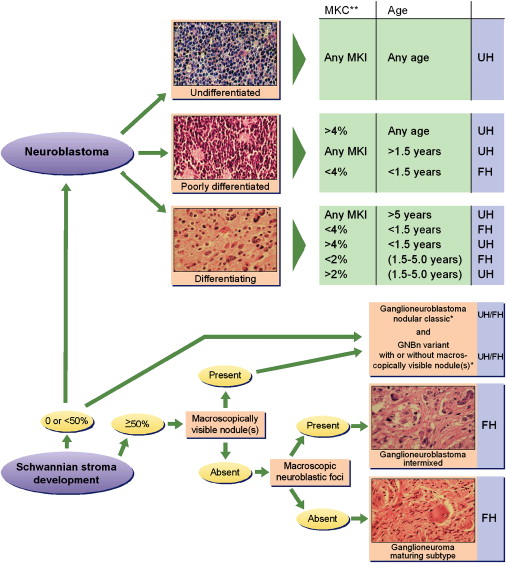
According to the INPC, the pNTs are assigned to one of the following four basic morphologic categories.
Neuroblastoma (Schwannian-stroma poor)
A neuroblastoma is a tumor composed of neuroblastic cells forming groups or nests separated by stromal septa with none to limited Schwannian proliferation. This category consists of the three subtypes: (1) undifferentiated, (2) poorly differentiated (background of recognizable neuropil and <5% of cells showing differentiation), and (3) differentiating (abundant neuropil and >5% cells showing differentiation toward ganglion cells).
Ganglioneuroblastoma, intermixed (Schwannian stroma-rich)
An intermixed ganglioneuroblastoma is a tumor containing well-defined microscopic nests of neuroblastic cells intermixed or randomly distributed in the ganglioneuromatous stroma. The nests are composed of a mixture of neuroblastic cells in various stages of differentiation, usually dominated by differentiating neuroblasts and maturing ganglion cells in a background of neuropil.
Ganglioneuroblastoma, nodular (composite Schwannian stroma-rich/stroma dominant and stroma-poor)
A nodular ganglioneuroblastoma is characterized by the presence of grossly visible, usually hemorrhagic neuroblastic nodules (stoma-poor component, representing an aggressive clone) co-existing with ganglioneuroblastoma, intermixed (stroma-rich component) or with ganglioneuroma (stroma-dominant component), both representing a nonaggressive clone. The term “composite” implies that the tumor is composed of biologically different clones.
Ganglioneuroma (Schwannian-stroma-dominant)
This variant has two subtypes: maturing and mature. The maturing subtype is composed predominantly of ganglioneuromatous stroma with scattered collections of differentiating neuroblasts or maturing ganglion cells in addition to fully mature ganglion cells. The mature subtype is composed of mature Schwannian stroma and ganglion cells.
There is a significant correlation between morphologic features of the INPC and biological properties of the pNTS, such as MYCN amplification or TrkA Expression.
Clinical presentation, diagnosis, and staging
Clinical presentation of neuroblastoma is dependent upon site of tumor origin, disease extent, and the presence of paraneoplastic syndromes. Neuroblastoma can arise anywhere along the sympathetic nervous system. The majority of tumors (65%) arise in the abdomen, with over half of these arising in the adrenal gland. Additional sites of origin include the neck, chest, and pelvis. There is a concordance with age and site of disease, with infants more likely to present with thoracic and cervical primary sites ( Fig. 2 ). One percent of patients have no detectable primary tumor.
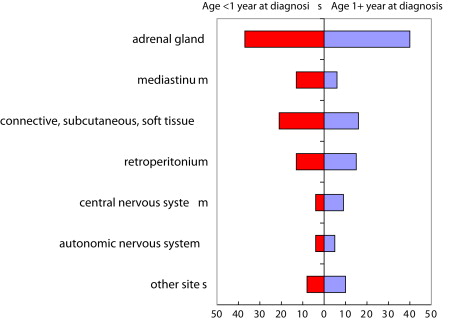
Approximately 50% of patients present with localized or regional disease, and approximately 35% of patients have regional lymph node spread at the time of diagnosis. Patients who have localized disease are often asymptomatic, with disease coincidently diagnosed after testing for unassociated medical conditions. Alternatively, mass or abdominal distension and pain are present. Patients who have localized cervical disease arising from the superior cervical ganglion may present with Horner syndrome. Epidural or intradural extension of tumor occurs in approximately 5% to 15% of patients diagnosed with neuroblastoma and may be accompanied by neurologic impairments. Well-documented paraneoplastic or clinical syndromes can be present at diagnosis and are summarized in Table 1 .
| Eponym | Syndrome Features |
|---|---|
| Pepper syndrome | Massive involvement of the liver with metastatic disease with or without respiratory distress. |
| Horner syndrome | Unilateral ptosis, myosis, and anhydrosis associated with a thoracic or cervical primary tumor. Symptoms do not resolve with tumor resection. |
| Hutchinson syndrome | Limping and irritability in young child associated with bone and bone marrow metastases. |
| Opsoclonus Mycoclonus Ataxia syndrome | Myoclonic jerking and random eye movement with or without cerebellar ataxia. Often associated with a biologically favorable and differentiated tumor. The condition is likely immune mediated, may not resolve with tumor removal, and often exhibits progressive neuropsychologic sequelae. |
| Kerner-Morrison syndrome | Intractable secretory diarrhea due to tumor secretion of vasointestinal peptides. Tumors are generally biologically favorable. |
| Neurocristopathy syndrome | Neuroblastoma associated with other neural crest disorders, including congenital hypoventilation syndrome or Hirshprung disease. Germline mutations in the paired homeobox gene PHOX2B have been identified in a subset of such patients. |
Disease dissemination occurs through lymphatic and hematogenous routes. Bone, bone marrow, and liver are the most common sites of hematogenous spread, with particular predilection for metaphyseal, skull, and orbital bone sites. In contrast to the frequent lack of symptoms with locoregional disease, patients who have widespread disease are often ill appearing with fever, pain, and irritability. A classic presentation of periorbital swelling and ecchymoses (“raccoon eyes”) is seen in children who have disease spread to periorbital region. Rarely, infants can present with respiratory compromise secondary to diffuse tumor involvement in the liver and massive hepatomegaly.
The current criteria for diagnosis and staging of neuroblastoma are based upon the International Neuroblastoma Staging System (INSS) criteria initially formulated in 1986 and revised in 1988. Neuroblastoma diagnosis is defined by pathologic confirmation from tumor tissue or by pathologic confirmation of neuroblastoma tumor cells in a bone marrow sample in the setting of increased urine or serum catecholamines or catecholamine metabolites (dopamine, vanillylmandelic acid, and homovanillic acid). Initial diagnostic testing should include CT or MRI to evaluate primary tumor size and regional extent and to assess for distant spread to neck, thorax, abdomen, or pelvic sites. Brain imaging is recommended only if clinically indicated by examination or neurologic symptoms. Bilateral posterior iliac crest marrow aspirates and core biopsies are required to exclude marrow involvement. Metaiodobenzylguanidine, a norepinephrine analog, is concentrated selectively in sympathetic nervous tissue and, when labeled with radioactive iodine (I 131 or I 123 ), is an integral component of neuroblastoma staging and response evaluation. A technetium bone scan should be considered for detection of cortical bone disease, especially in patients who have a negative metaiodobenzylguanidine scan.
The INSS definitions for neuroblastoma stage are listed in Table 2 . Resectability implies tumor removal without removal of vital organs, compromise of major vessels, or patient disfigurement. Completely resected tumors are classified as stage 1, and partially resected regional tumors with or without regional nodal involvement are classified as stages 2 and 3 dependent upon amount of tumor resection, local invasion, and regional lymph node involvement. Stage 4 disease is defined as distant nodal or hematogenous spread of disease. A unique pattern of dissemination limited to liver, skin, and minimal bone marrow involvement has been described in infants (stage 4s), which has a potential for spontaneous regression in marked contrast to the disseminated aggressive disease seen in the majority of patients greater than 18 months of age.
| Stage 1 | Localized tumor with complete gross excision with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically (nodes attached to and removed with the primary tumor may be positive) |
| Stage 2A | Localized tumor with incomplete gross resection; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
| Stage 2B | Localized tumor with or without complete gross excision with ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically |
| Stage 3 | Unresectable unilateral tumor infiltrating across the midline a with or without regional lymph node involvement, localized unilateral tumor with contralateral regional lymph node involvement, or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement b |
| Stage 4 | Any primary tumor with dissemination to distant lymph nodes, bone, bone marrow, liver, skin, or other organs (except as defined for stage 4S) |
| Stage 4S | Localized primary tumor (as defined for stage 1, 2A, or 2B) with dissemination limited to skin, liver, or bone marrow c (limited to infants <1 yr of age) |
a The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
b Proven malignant effusion within the thoracic cavity if it is bilateral or the abdominal cavity upstages the patient to INSS stage 3.
c Marrow involvement in stage 4S should be minimal (ie, <10% of total nucleated cells identified as malignant on bone marrow biopsy or marrow aspirate). More extensive marrow involvement would be considered to be stage 4. The metaiodobenzylguanidine scan (if performed) should be negative in the marrow.
Tumor biology and prognosis
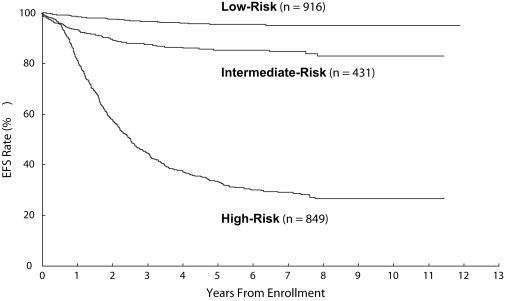
Numerous clinical and biologic factors have been shown to predict clinical behavior of neuroblastoma. There is international agreement that a combination of clinical and biologic factors best predicts clinical prognosis. The Children’s Oncology Group stratifies patients into low-, intermediate-, or high-risk categories based upon age at diagnosis, INSS stage, tumor histopathology, DNA index (ploidy), and MYCN amplification status ( Table 3 ), with each group displaying a unique risk for recurrence ( Fig. 3 ). Similar strategies are used internationally; however, several factors outlined below suggest a potential for continued evolution of any classification algorithm.
| Risk Group | Stage | Age | MYCN Amplification Status | Ploidy | Shimada |
|---|---|---|---|---|---|
| Low risk | 1 | Any | Any | Any | Any |
| Low risk | 2a/2b | Any | Not amplified | Any | Any |
| High risk | 2a/2b | Any | Amplified | Any | Any |
| Intermediate risk | 3 | <547 d | Not amplified | Any | any |
| Intermediate risk | 3 | ≥547 d | Not amplified | Any | FH |
| High risk | 3 | Any | Amplified | Any | Any |
| High risk | 3 | ≥547 d | Not amplified | Any | UH |
| High risk | 4 | <365 d | Amplified | Any | Any |
| Intermediate risk | 4 | <365 d | Not amp | Any | Any |
| High risk | 4 | 365 to <547 d | Amplified | Any | Any |
| High Risk | 4 | 365 to <547 d | Any | DI = 1 | Any |
| High risk | 4 | 365 to <547 d | Any | Any | UH |
| Intermediate risk | 4 | 365 to <547 d | Not amplified | DI > 1 | FH |
| High risk | 4 | ≥547 d | Any | Any | Any |
| Low risk | 4s | <365 d | Not amplified | DI > 1 | FH |
| Intermediate risk | 4s | <365 d | Not amplified | DI = 1 | Any |
| Intermediate risk | 4s | <365 d | Not amplified | Any | UH |
| High risk | 4s | <365 d | Amplified | Any | Any |
Stage and Age
Although the prognostic value of classifying patients according to the INSS has been confirmed, an inherent bias toward complete resection exists. Variation in the surgical approach for resection of locoregional disease has the potential to dramatically alter INSS staging. Such variation has led to considerations of alternative staging approaches. In 1995, the European International Society of Pediatric Oncology Neuroblastoma group demonstrated that radiographic characteristics of the tumor, termed surgical risk factors, were useful in predicting the ability to resect the primary tumor and the risk of developing postoperative complications. This led to proposing an International Risk Group imaging classification system that would use surgical risk factors to more uniformly define extent of disease/staging. The prognostic significance of the International Risk Group classification system will be prospectively validated in ongoing trials within the United States and Europe and, if validated, may replace the current INSS staging.
Age is an important clinical prognostic factor. Patients older than 1 to 2 years have a worse prognosis than those who are younger, especially for patients who have disseminated disease at diagnosis. Until recently, an age of 365 days has been used as a surrogate for tumor behavior, although alternative ages have been explored. A review of over 3000 neuroblastoma cases confirmed age as a continuous prognostic variable and identified 460 days as the most prognostic age cutoff. Recent reviews demonstrated that patients up to 18 months of age who are diagnosed with biologically favorable INSS stage 3 and 4 neuroblastoma share the same excellent prognosis with those less than 1 year of age and suggests that 18 months may be a more clinically relevant predictor of outcome. These outcomes were achieved with dose-intensive chemotherapy regimens, including myeloablative consolidation therapy as compared with the moderately dosed chemotherapy received by the more biologically favorable patient cohorts. The ability to treat children ages 12 to 18 months of age who have metastatic disease or locoregional disease using less aggressive therapy is under investigation.
Pathology
The pathologic characteristics of neuroblastoma have been used to further classify these tumors as outlined previously. Shimada initially proposed a histology-based classification of tumors into “favorable” and “unfavorable” by combining age with extent of tumor differentiation, presence of a Schwannian stromal components, and degree of mitosis and karyorrhexis. The prognostic impacts of the original Shimada classification and the more recently revised INPC have been validated. The inclusion of age, which is a strong independent prognostic variable, has led to consideration for examining cellular proliferation and extent of differentiation as independent variables in future multifactorial clinical and biological classification schemas.
Genetic Factors
Neuroblastoma can be divided into those with a near-diploid nuclear DNA content (≈45%) and near-triploid tumors (≈55%). Near-triploid neuroblastomas are characterized by whole chromosome gains and losses without structural genetic aberrations. Clinically, near-triploid tumors are more often localized and show a favorable outcome. Near-diploid neuroblastomas are characterized by the presence of genetic aberrations, such as MYCN amplification, 17q gain, and chromosomal losses (reviewed in references ). For many genetic, molecular, and clinical factors, a prognostic value has been reported. Recently, a systematic review of prognostic tumor markers in neuroblastoma has been published. Riley and coworkers identified 3415 papers with a sensitive literature search for prognostic factors in neuroblastoma, of which 428 were judged to be relevant. In total, 31 prognostic factors were reported, each in five or more papers. Among these 31 prognostic factors, there were six genetic aberrations listed (ploidy, MYCN amplification, chromosome 1 p loss, chromosome 17q gain, chromosome14q loss, and loss of chromosome 11q). Meta-analysis of prognostic markers showed that MYCN amplification and DNA ploidy had the strongest prognostic impact. The pooled hazard ratio for a bad outcome (measured by overall survival) for MYCN amplification was 5.48 (95% confidence interval, 4.30–6.97) and for DNA near-diploidy was 3.23 (95% confidence interval, 2.08–5.00).
Most neuroblastomas have a nuclear DNA content in the diploid range. Tumors from patients who have lower stages of disease can often be hyperdiploid or near-triploid (reviewed in references ). DNA content is most prognostic in infants who have neuroblastoma. The Children’s Oncology Group is the only collaborative group using ploidy of diagnostic tumor specimen for risk stratification (see Table 3 ).
Allelic Gains and Amplifications
The MYCN oncogene is present in an increased copy number in 25% to 35% of neuroblastomas. MYCN amplification is found in 30% to 40% of stage 3 and 4 neuroblastomas and in only 5% of localized or stage 4s neuroblastomas. MYCN -amplified neuroblastomas are characterized by a highly aggressive behavior with unfavorable outcome (reviewed in ). Some discussion remains on the prognostic value of MYCN amplification in the rare cases of completely resected localized neuroblastoma. The unfavorable prognostic value also holds in the prognostically favorable group of infants who have stage 4s neuroblastoma. In MYCN -amplified neuroblastomas, loss of chromosome 1 p is almost invariably present.
Allelic Gains
Gain of the entire chromosome 17 or gain of parts of chromosome 17q occur in greater than 60% of neuroblastomas. The partial 17q gain most often results from unbalanced translocation of 17q21–25 to another chromosome (eg, chromosome 1). Partial gain of 17q identifies unfavorable neuroblastoma. Obvious candidate genes on 17q are the NM23 and the BIRC5 (survivin) gene.
Tumor Suppressor Genes
Loss of tumor suppressor regions is reported in neuroblastomas for many chromosomal regions. The most frequently affected regions are chromosome 1 p (30–40%), 4 p (20%), 1lq (25%), and 14q (25%).
Chromosome 1 p loss occurs more frequently in older children who have stage 3 and 4 neuroblastoma and is correlated with increased serum ferritin and serum lactate dehydrogenase. In almost all samples with MYCN amplification, concomitant 1 p loss is demonstrated, but loss of chromosome 1 p also occurs in MYCN single-copy cases. Several studies have shown that 1 p loss is a strong predictor of outcome. In the meta-analysis of Riley and colleagues, MYCN amplification and ploidy were stronger predictors of outcome, indicating that 1 p loss might best be used to identify high-risk patients in MYCN single-copy tumors.
Chromosome 11q loss is demonstrated in approximately 40% of patients (reviewed in ). Chromosome 11q loss is inversely correlated with MYCN amplification and therefore identifies an additional high-risk subset of patients characterized by advanced stage, older age, and unfavorable pathology. It has recently become clear that “unbalanced” deletion of 11q (deletion of long-arm material with retention or gain of short-arm material) occurs in 15% to 20% of cases but is more clearly associated with high-risk biologic features. Prospective evaluation of 1 p and 11q status is ongoing in several cooperative group trials and is especially aimed at risk stratification of intermediate-risk patients.
Molecular Factors
Despite extensive data correlating genomic alterations with disease outcome, no bona fide target genes have been identified for neuroblastoma, with the exception of MYCN . A number of biological pathways regulating cancer seem to be disrupted or affected in neuroblastoma, including tumor differentiation, apoptosis, drug resistance, angiogenesis, and metastasis. Insight into the molecular regulation of these biological pathways will lead to the identification of novel drug targets.
Because neurotrophin signaling has a central role in normal neuronal development and may be involved in differentiation and regression of neuroblastoma there has been interest in alterations of these pathways. The clinical and biological roles of Trk receptors ( NTRK1 , NTRK2 , and NTRK3 encoding TrkA, TrkB, and TrkC, respectively) and their ligands (NGF, BDNF, and NT-3, respectively) have been extensively investigated. Trk-receptors have been identified as important prognostic factors in neuroblastoma. High expression of TrkA is present in neuroblastomas, with favorable biological features and correlates with good outome. By contrast, full-length TrkB is highly expressed in biologically unfavorable, MYCN -amplified, aggressive neuroblastomas. In neuroblastoma cell culture models, the biological effects of TrkA include neuronal differentiation or apoptosis, depending on the presence or absence of NGF and on inhibition of proliferation and angiogenesis. TrkA signaling might also be related to Phox2B- or Delta-Notch–regulated differentiation programs. Depending on the microenvironment, NGF/TrkA signaling could provoke differentiation or regression in favorable neuroblastomas. This would partly explain spontaneous regression as a delayed activation of developmentally programmed cell death resulting from the absence of NGF in the microenvironment.
Activation of TrkB by its ligand BDNF results in enhanced proliferation, migration, angiogenesis, and chemotherapy resistance of neuroblastoma cells. Differential splicing of Trk receptors results in the expression of truncated receptors lacking the kinase domain, which may function as dominant negative inhibitors or scavenger receptors sequestering the ligand. Truncated TrkB seems to be preferentially expressed in more differentiated tumors. Recently, a novel NGF-unresponsive TrkA splice variant has been identified, which is predominantly expressed in clinically aggressive neuroblastomas. This splice variant promotes cell survival, xenograft tumor growth, and angiogenesis. Thus, the neurotrophins and their receptors govern numerous cellular functions in neuroblastoma, and the complexity of the signaling responses is reflected in the multiplicity of the pathways involved. More detailed insights into the mechanisms regulating differentiation might suggest new options for treatment.
Delayed activation or disruption of normal apoptotic pathways may be an important phenomenon involved in spontaneous regression and therapy resistance of neuroblastoma. Major elements of the apoptotic signaling cascade with abnormal expression or activation patterns include the BCL2 family, survivin, and caspase-8. The latter is mainly affected by inactivation due to epigenetic silencing. CpG-island hypermethylation of gene promoters is a frequent mechanism for functional inactivation of genes. In neuroblastoma, this mode of inactivation has been demonstrated not only for caspase-8 but also for the four TRAIL apoptosis receptors, the caspase-8 inhibitor FLIP, the RASSF1A tumor suppressor, p73, RB1, DAPK, CD44, p14ARF, and p16INK4a. Because many of these genes are involved in apoptotic signaling and therapy responsiveness, gene hypermethylation might be a major event leading to resistance. Therefore, the antitumor effects of demethylating agents, including decitabine, are being investigated in preclinical studies.
Acquired resistance to chemotherapeutic agents may be conferred by enhanced drug efflux due to overexpression of classical multidrug resistance proteins, including multidrug resistance gene 1 and the gene for multidrug resistance-related protein. Their potential clinical significance in neuroblastoma has been addressed in several studies, but their interaction with each other and the role of unknown cofactors remains to be elucidated. Several additional factors have been shown to contribute to treatment resistance in neuroblastoma, including expression of oncogenes such as MYCN, TrkB/BDNF signaling, or loss of p53 expression.
Enhanced tumor angiogenesis and high expression of proangiogenic factors such as vascular endothelial growth factor and basic fibroblast growth factor are correlated with an aggressive phenotype in neuroblastoma, making angiogenesis inhibitors an attractive treatment option that is being evaluated in preclinical studies. Despite the fact that approximately 50% of patients present with disseminated disease at the time of diagnosis, little is known about the biology of invasion and metastases in neuroblastoma. Major molecular players in the regulation of local invasiveness and metastases are metalloproteinases (mainly MMP9), activating matrix-degrading proteolytic enzymes, and molecules regulating tumor cell adhesion and migration, such as CD44 and NM23-H1.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


