Neuroblastoma
Garrett M. Brodeur
Michael D. Hogarty
Rochelle Bagatell
Yael P. Mossé
John M. Maris
INTRODUCTION
Few tumors have engendered as much fascination and frustration for clinicians and researchers as neuroblastoma, the most common and deadly solid tumor of childhood. These tumors may regress spontaneously, particularly in infants, or they may mature into a benign ganglioneuroma. In contrast, many older children have unresectable or metastatic disease at the time of diagnosis, and their overall prognosis has been poor. However, genomic and biologic analysis of tumor tissue has identified distinct differences in genotype and expression patterns that shed considerable light on these disparate clinical behaviors.
Indeed, we have entered a new era in terms of our understanding of the genetic and genomic changes that underlie neuroblastoma predisposition and pathogenesis. Two genes that are responsible for most cases of hereditary neuroblastoma have been identified. Genome-wide association studies have discovered additional genes that also contribute to neuroblastoma susceptibility and pathogenesis. Exome-based and complete genomic sequencing of tumor DNA obtained at diagnosis has revealed recurrent mutations in a relatively low number of genes, emphasizing the importance of epigenetic regulation in this embryonal cancer. Together, new rational targets for therapeutic intervention have been identified, highlighting the potential for personalized approaches to disease management.
Patterns of genetic change allow neuroblastomas to be classified into subsets with distinct genomic/biologic features and clinical behavior. Indeed, certain genomic abnormalities are very powerful predictors of response to therapy and outcome, and they have become essential components of tumor characterization at diagnosis. Thus, neuroblastoma serves as a model tumor in which the genomic and biologic analyses of tumor cells provide important information that is already guiding optimal patient management. The challenge for the next decade is to translate this information into more effective and less toxic therapy for these patients. This chapter reviews the current understanding of various clinical and biologic features of neuroblastoma and relates these characteristics to trends in predicting outcome as well as future strategies for targeted treatment. It also reviews the current approach to the diagnosis, staging, and management of patients with neuroblastoma.
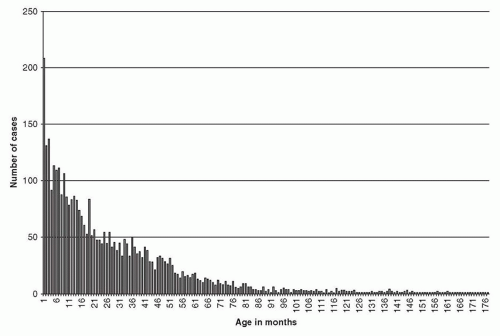 Figure 30.1 Distribution of age at diagnosis for 3,666 neuroblastoma patients. The age at diagnosis in months (x-axis) and the number of patients at that age (y-axis) are shown. (Courtesy of W.B. London, COG Statistical Office.) |
INCIDENCE AND EPIDEMIOLOGY
Incidence
Neuroblastoma is the most common extracranial solid tumor in children, accounting for 7% to 8% of all childhood cancers. The prevalence is about 1 case per 7,000 live births, and there are about 800 new cases of neuroblastoma per year in the United States.1 This corresponds to an incidence of 9.7 per million per year in white children and 6.8 per million per year in black children younger than 19 years.1 Evidence indicates that this incidence is fairly uniform throughout the world, at least for industrialized nations.2 Neuroblastoma is slightly more common in boys than in girls, with a male-to-female ratio of 1.1 to 1 in most large studies. Neuroblastoma is the most common cancer diagnosed during infancy.1 Review of 3,666 neuroblastoma patients registered in cooperative group studies at Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG) institutions from 1986 to 2001 showed a median age at diagnosis of about 19 months. In this cohort, 36% were infants, 89% were younger than 5 years, and 98% were diagnosed by 10 years of age3 (Fig. 30.1). The distribution of cases by age clearly shows that this is a disease of infancy and early childhood, with the highest number of cases diagnosed in the first month of life.
Epidemiology
The etiology of neuroblastoma is unknown, and to date there are no data supporting a major role for environmental exposures. No particular prenatal or postnatal exposure to drugs, chemicals, viruses, or radiation has been associated strongly, consistently, or unequivocally with an increased or decreased incidence of neuroblastoma.4 Likewise, no features have been consistently associated with a protective effect, such as maternal vitamin use or early childhood infections and allergies. However, replication of association in a rare disease is often difficult, and the data available do not preclude gene-environment interactions in neuroblastoma. There has been an association between congenital anomalies, especially cardiac or urogenital anomalies, and neuroblastoma.5,6 These cases may reflect some underlying genetic abnormality that contributes to both, but no consistent pattern has been identified (see below).
Mass-Screening Studies
The outcome of infants with neuroblastoma is substantially better than that of older children with this disease. Furthermore, almost all neuroblastomas produce catecholamines, and their metabolites, homovanillic acid (HVA) and vanillylmandelic acid (VMA), can be readily detected in urine. Therefore, mass screening of infants for neuroblastoma by measuring urine VMA and HVA was initiated in Japan with the goal of detecting tumors earlier. Early results were promising, so similar efforts were initiated in North America (Quebec, Canada) and in Europe (France and Germany).7,8 Current results indicate that mass screening has resulted in a substantial increase in the prevalence of, neuroblastoma in screened compared with unscreened populations, but without a significant decrease in the prevalence of or mortality due to, neuroblastoma in patients older than 1 year.9,10,11 Thus, the goal to reduce neuroblastoma mortality by mass screening in infancy was not achieved, and these efforts have essentially stopped throughout the world.
Nevertheless, mass-screening studies have provided valuable insights into the pathogenesis and clinical behavior of biologically favorable tumors. The increased prevalence of neuroblastoma that was observed in screened populations suggests that spontaneous regression of neuroblastoma (without clinical detection) is at least as prevalent as neuroblastoma that is detected clinically. In addition, analyses performed on screened tumors show that virtually all of them, regardless of stage, were genomically and biologically favorable.12,13 This is in contrast to the higher prevalence of unfavorable biologic features found in older children that were detected clinically. Importantly, these studies also suggested that biologically unfavorable tumors did not arise from these biologically favorable tumors. Recent advances in identifying neuroblastoma predisposition and susceptibility genes may identify individuals who are at increased risk for neuroblastoma, and future screening efforts should be focused on them.
GENETIC PREDISPOSITION
Like most human cancers, a subset of patients with neuroblastoma exhibits a predisposition to develop this disease, and this predisposition follows an autosomal dominant pattern of inheritance, with high but variable penetrance.14 A family history of neuroblastic tumors is found in about 1% to 2% of newly diagnosed cases, with a standardized incidence ratio of 9.7 for siblings of index cases.15 Neuroblastoma pedigrees show notable heterogeneity in the type of tumors that arise, with both benign and malignant forms occurring in the same family.16 Patients with hereditary neuroblastoma differ from those with sporadic disease, in that they are diagnosed at an earlier age and/or with more than one primary tumor, clinical characteristics that are hallmarks of cancer predisposition syndromes. There is incomplete penetrance, which may be accounted for, in part, by spontaneous regression following malignant transformation in a subset of cases. Genome-wide linkage scans in neuroblastoma pedigrees discovered activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) oncogene in 75% to 80% of familial cases.17,18,19,20 These germline mutations encode for single-base substitutions in key regions of the kinase domain that result in constitutive activation of the kinase and a premalignant state. Similar activating mutations are also somatically acquired in approximately 10% of sporadic neuroblastomas (see below).17,18,19,20
Neuroblastoma occasionally occurs in association with disorders related to abnormal development of neural-crest-derived tissues, including congenital central hypoventilation syndrome and Hirschsprung disease.21 Missense, nonsense, and polyalanine repeat expansion mutations in PHOX2B were shown to be responsible for these heritable disorders and accounted for approximately 5% of hereditary neuroblastomas.22,23 PHOX2B is a homeobox gene that is a master regulator of normal autonomic nervous system development, and inactivating mutations of this gene account for this rare field defect of sympathoadrenal tissues.
Gain-of-function mutations in ALK or inactivating mutations of PHOX2B account for 80% to 85% of hereditary neuroblastomas. Therefore, genetic testing for mutations in these two genes should be considered whenever a patient has a family history of the disease or evidence for multiple primary tumors (e.g., bilateral adrenal tumors) suspicious for a highly penetrant transmissible mutation. If mutations are found, this should be followed by appropriate genetic counseling and cancer surveillance protocols. However, there are another 15% to 20% of hereditary neuroblastoma cases that are not accounted for by these mutations, suggesting that one or more additional predisposition genes remain to be discovered.
Constitutional chromosome abnormalities are rare in patients with neuroblastoma, but these rare cases may help localize genes critical to tumor initiation.24 Patients frequently show associated congenital disorders, and these may be quite severe. Interestingly, constitutional deletions and/or unbalanced translocations may occur at loci known to be rearranged somatically in tumor tissues, such as 1p, 2p (MYCN and ALK), 11q, and 17q.14,24,25,26,27,28,29 These rare cases further emphasize the complex underlying genetics of neuroblastoma and suggest that aberrant expression or regulation of multiple genes may work together to initiate malignant transformation of undifferentiated sympathoadrenal neuroblasts.
The vast majority of neuroblastoma cases occur sporadically. For many of these patients, malignant transformation may arise from a synergistic interaction of common DNA variations, with each individual variation having a relatively modest impact on susceptibility. A large genome-wide association study has identified dozens of polymorphic alleles across the genome that are strongly associated with neuroblastoma susceptibility.30,31,32,33,34,35,36 These loci generally are localized in regulatory regions (e.g., enhancer elements) that influence genes involved in sympathetic nervous system development. These polymorphisms not only influence disease susceptibility, but also the phenotype that arises, because certain polymorphisms are associated primarily with either low- or high-risk neuroblastoma.31,33,34,37 These data further emphasize the profound influence of the heritable genome on susceptibility to cancer, as well as how the disease evolves once initiated.
MOLECULAR PATHOGENESIS
Germline ALK or PHOX2B mutations predispose to neuroblastoma with high penetrance, credentialing these as bona fide neuroblastoma driver genes. In other patients, the inheritance of common DNA variants associated with increased neuroblastoma risk may contribute modestly to tumor susceptibility. However, the overwhelming majority of children presenting with this tumor do
not have a strong genetic predisposition or family history of neuroblastoma. Tumor initiation in these sporadic cases is a consequence of somatically acquired genomic changes, presumably in these or other oncogenic drivers. Many such recurrent genomic changes have been identified and correlated with tumor behavior, and these prognostic factors are used in risk classification schemas worldwide. More recently, exome-wide and whole-genome sequencing of diverse neuroblastomas has provided base-pair resolution of somatic mutations. Although such efforts have led to the identification of additional putative driver genes, the most striking finding is the overall paucity of mutations and the relative absence of recurrently mutated genes, suggesting a strong role for epigenetic regulation of gene expression in the initiation and progression of neuroblastoma and other pediatric neoplasms.
not have a strong genetic predisposition or family history of neuroblastoma. Tumor initiation in these sporadic cases is a consequence of somatically acquired genomic changes, presumably in these or other oncogenic drivers. Many such recurrent genomic changes have been identified and correlated with tumor behavior, and these prognostic factors are used in risk classification schemas worldwide. More recently, exome-wide and whole-genome sequencing of diverse neuroblastomas has provided base-pair resolution of somatic mutations. Although such efforts have led to the identification of additional putative driver genes, the most striking finding is the overall paucity of mutations and the relative absence of recurrently mutated genes, suggesting a strong role for epigenetic regulation of gene expression in the initiation and progression of neuroblastoma and other pediatric neoplasms.
Patterns of Somatic Genomic Changes
Early karyotype analyses revealed most neuroblastomas to be aneuploid, with alterations that include genomic amplifications, unbalanced translocations, and whole chromosome and segmental chromosome losses and gains. Subsequent molecular studies have validated and extended these findings for specific genomic sites, many of which retain strong prognostic value and are used by groups worldwide in risk classification and therapy planning.38,39,40,41 The accumulated research on the genomics and biology of neuroblastoma provides for an evolutionary model that proposes that all neuroblastomas arise from a common precursor, but that different types of genomic instability lead to different patterns of alterations, leading to divergent clinical behaviors.38,39,40,41,42 The model predicts two major subtypes that are biologically distinct, in that unfavorable tumors do not evolve or progress from favorable tumors, and vice versa42(Fig. 30.2).
Type 1
Type 1 neuroblastomas are characterized by gains and losses of whole chromosomes, with few if any segmental (or partial) chromosome alterations.39,42 Defective mitotic segregation likely contributes, although the causative mechanisms are unknown. These tumors express high levels of TrkA43,44,45 (see receptor tyrosine kinases, below) and are prone to differentiation or apoptosis, which may be nerve growth factor (NGF) dependent. They tend to have favorable biologic and clinical features, including lower stage and younger age at diagnosis (<18 months), and the prognosis for such children is excellent. Their hyperdiploid (often near-triploid) modal karyotype reflects primarily whole chromosome gains, and they lack MYCN amplification or other specific, segmental chromosome alterations that are associated with aggressive behavior. Hyperdiploid DNA content as determined by flow cytometry remains a favorable prognostic biomarker in children under 18 months of age with stage 4 or 4S disease, as it is highly predictive of favorable disease outcome.46,47,48 Hyperdiploidy may also have favorable prognostic value for localized tumors with MYCN amplification.49
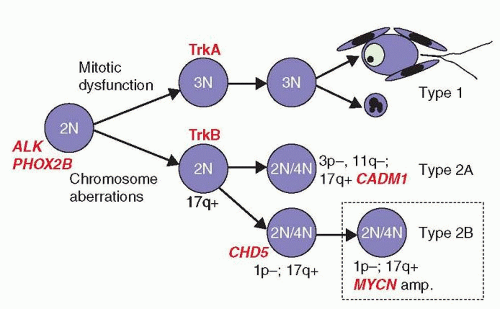 Figure 30.2 Genetic model of neuroblastoma development (see text). (Modified from Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003;3(3):203-216.) |
Type 2
Type 2 tumors are characterized by segmental chromosome alterations, often arising from unbalanced deletions or translocations, with or without additional whole chromosome changes.39,42 Segmental alterations are recurrently identified within chromosome arms 1p, 1q, 3p, 11q, 14q, and/or 17q, although they may occur elsewhere. Tumors in this group tend to have a near-diploid or near-tetraploid karyotype, as the segmental alterations do not markedly alter total DNA content. Two further subtypes can be distinguished: the first (type 2A) is characterized by segmental deletions at 3p, 11q, and other chromosomes in tumors that do not have MYCN amplification or 1p deletion; the second (type 2B), comprising the most aggressive subset, has amplification of the MYCN oncogene usually accompanied by 1p deletion and additional segmental alterations, but usually without loss of 11q or 3p. These tumors frequently have unbalanced 17q gain, and they usually express the TrkB receptor plus brain-derived neurotrophic factor (BDNF) ligand, which presumably activates an autocrine survival pathway to confer a selective advantage.50
Although full genomic characterization of all tumors at presentation may be a realizable goal in the future, it is clear that the detection of a subset of prognostic segmental chromosome alterations can be integrated with traditional clinical factors to refine neuroblastoma risk classification. A number of such specific molecular features are discussed below.
Oncogenic Driver Genes in Neuroblastoma
MYCN
Neuroblastoma karyotypes frequently reveal the cytogenetic hallmarks of gene amplification, namely, double-minute chromosomes (DMs) or homogeneously staining regions (HSRs). Schwab and colleagues originally identified the MYC-related oncogene MYCN (2p24) as the target of this amplification event,51,52 although additional genes may be coamplified.53,54 Genomic amplification events independent of MYCN are rare.39,55 Brodeur and Seeger first showed that amplification of MYCN is associated with advanced stages of disease, unfavorable biologic features, and a poor outcome56,57 (Table 30.1). Indeed, it is also independently associated with poor outcome in otherwise favorable patient groups, underscoring its biologic importance.47,56,57,58 Currently, the MYCN gene status in tumors is routinely determined at diagnosis to assist in therapy planning. Interphase fluorescence in situ hybridization (FISH) is the technique of choice because verification of the MYCN signal affords outstanding quality control, while also allowing detection of low-level amplification and intratumoral
heterogeneity59,60 (Fig. 30.3). The clinical relevance of these less common findings is being evaluated prospectively.
heterogeneity59,60 (Fig. 30.3). The clinical relevance of these less common findings is being evaluated prospectively.
TABLE 30.1 Relationship of MYCN Amplification with Extent of Disease at Diagnosis | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
The degree of increased copy number required to constitute “amplification” remains arbitrary but most groups define this as >4 times the number of MYCN copies (FISH signals) as compared with a control 2p probe. In most tumors with MYCN amplification, the MYCN copy number greatly exceeds this threshold (50 to 400 copies/cell). The overall prevalence of MYCN amplification is about 20% to 22%,42 with near-complete concordance when multiple sites are assessed, or between samples obtained at diagnosis and relapse.61 Therefore, it is an inherent feature of a subset of aggressive tumors at diagnosis, rather than a mutation acquired during tumor progression. In general, levels of MYCN expression in tumors with amplification are much higher than in tumors without amplification.62 However, it is controversial whether or not “overexpression” of MYCN mRNA or MycN protein has prognostic significance independent of MYCN amplification.63,64
MYCN is a member of the MYC proto-oncogene family, which encodes transcription factors that regulate the expression of approximately 15% of all human genes, such that overexpression markedly impacts cell behavior.65,66 MYC genes play a large role in regulating metabolism and cellular biomass in support of proliferation and are commonly deregulated in diverse cancers. The most compelling evidence that deregulation of MYCN is a fundamental event in a subset of human neuroblastomas comes from a genetically engineered murine model of the disease. Weiss and coworkers67 directed MYCN expression to the murine neural crest using a tyrosine hydroxylase (TH) promoter. These animals frequently develop tumors of the sympathetic nervous system that resemble human neuroblastomas both morphologically and genomically.68 This mouse model is useful for understanding genetic events that cooperate with MycN protein overexpression to result in an aggressive malignant phenotype. This genetically engineered mouse model may also be useful to test new therapeutic strategies in a preclinical setting.
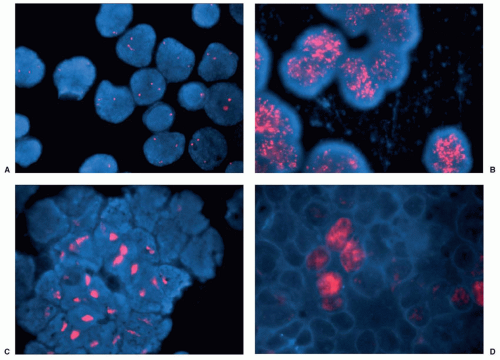 Figure 30.3 Detection of MYCN amplification by FISH in human neuroblastomas. Four panels of neuroblastoma cells hybridized with a fluorescent MYCN probe, and the nuclei counterstained with DAPI, are shown. A: Neuroblastoma cells without MYCN amplification, showing two to three discrete copies per cell. B: Neuroblastoma cells with MYCN amplification in the form of extrachromosomal DMs. The copy number is variable from cell to cell, and the extra copies are dispersed throughout the nucleus. C: Neuroblastoma cells with MYCN amplification in the form of chromosomally integrated homogeneously staining regions. The extra MYCN signal is more uniform from cell to cell, and the signal is localized to a discrete region of the nucleus. D: Focal MYCN amplification in a subset of the neuroblastoma cells, demonstrating heterogeneity in this cell population. (Courtesy of Lisa Moreau and A. Thomas Look at the Dana Farber Cancer Institute, Boston, MA.) |
ALK
The ALK gene encodes a cell surface receptor tyrosine kinase (RTK) expressed at significant levels only in the developing nervous system.69 Germline ALK mutations are the major cause of hereditary neuroblastoma.17,18,19,20 These mutations are single amino acid substitutions in the tyrosine kinase domain and promote constitutive signaling activity. This provides the first example of a germline mutation in an oncogene causing a pediatric cancer. Similar somatically acquired ALK-activating mutations are also found as oncogenic drivers in 8% to 10% of sporadic neuroblastomas.17,18,19,20 ALK gene amplification (of a wild-type ALK gene) accompanies MYCN amplification in another 2% to 4% of neuroblastomas, and imparts an oncogenic dependency.19,20 Therefore, 10% to 14% of neuroblastomas will have an activating ALK
alteration that may serve as a novel therapeutic target. ALK fusion proteins that arise through chromosomal translocations are found in anaplastic large cell lymphomas, non-small cell lung cancers, and other cancers. Crizotinib is an ALK inhibitor that has already earned approval for use in ALK-rearranged cancer and is being tested in neuroblastoma.70
alteration that may serve as a novel therapeutic target. ALK fusion proteins that arise through chromosomal translocations are found in anaplastic large cell lymphomas, non-small cell lung cancers, and other cancers. Crizotinib is an ALK inhibitor that has already earned approval for use in ALK-rearranged cancer and is being tested in neuroblastoma.70
Chromatin-Remodeling Genes
Whole-genome and exome-based sequencing have identified recurrent mutations in numerous chromatin-remodeling genes in diverse human cancers. The alpha-thalassemia/mental retardation syndrome X-linked gene, ATRX, was identified as recurrently mutated in neuroblastoma.71 This protein is part of a multiprotein Swi/Snf-like complex that regulates ATP-dependent chromatin-remodeling, nucleosome assembly, and telomere maintenance. Its germline inactivation leads to α-thalassemia and mental retardation but not to neuroblastoma or other tumors. However, somatic inactivating mutations have been identified in ATRX in neuroblastomas, and there is a striking association with older patient age at diagnosis. MYCN amplification is rare in tumors in older patients, and to date ATRX mutation and MYCN amplification remain exclusive. The functional impact of mutant ATRX on the cancer phenotype is unknown, but it may work via the alternative lengthening of telomeres pathway.
The ARID1A and ARID1B genes have also been identified as recurrently mutated in a subset of highest-risk neuroblastomas.72 These genes are mutually exclusive members of the Swi-Snf Brg1-associated factors (BAF) complex that is essential for the self-renewal of multipotent neural stem cells.73 It is postulated that these mutations disrupt the differentiation switch from neural progenitor to mature neuron that is mediated by intact BAF complex signaling. Like other bona fide neuroblastoma drivers MYCN and ALK, ARID1 gene mutations are overrepresented in cell lines, which represent the most aggressive subset of tumors. Indeed, loss-of-function mutations in ARID1A or ARID1B were associated with exceptionally poor outcome.72 ARID1A maps to chromosome 1p35.3 and is biallelically inactivated, suggesting a classic tumor suppressor role. ARID1B mutations (6q25), in contrast, are monoallelic with a retained wild-type allele. A more accurate assessment of the prevalence of ARID1A and ARID1B mutations awaits study in a larger cohort but is estimated to be 2% to 3% overall.
Recurrent Segmental Chromosome Alterations
Unbalanced gain of the long arm of chromosome 17 (17q) occurs in over half of all neuroblastomas.74 Although gain of 17q can occur independently, it frequently occurs as part of an unbalanced translocation between chromosomes 1 and 17 with preferential gain of a region from 17q22-qter.75,76 The genes responsible for the selective advantage are unknown, but BIRC5 (survivin, an inhibitor of apoptosis) and NME1 (NM23, involved in the synthesis of nucleoside triphosphates) have been postulated as targets.77,78 Unbalanced gain of 17q is associated with more aggressive neuroblastomas, although its independent prognostic significance awaits a large prospective trial and multivariate analysis. In contrast, gain of whole chromosome 17 is characteristic of the hyperdiploid favorable neuroblastoma subtype.39
Deletion of the short arm of chromosome 1 (1p) is found in about 35% of primary neuroblastomas, and it is associated with advanced stages of disease and MYCN amplification.79,80,81,82,83 Allelic loss at 1p36 also independently predicts for an increased risk of relapse in patients with localized tumors and tumors lacking MYCN amplification.79,84,85 A number of laboratories have tried to identify the tumor suppressor gene(s) deleted from this region by looking for homozygous deletion, but such events are extremely rare.86,87,88 Brodeur and coworkers89 defined a <2 Mb smallest region of consistent deletion after examining over 1,200 neuroblastomas and identified the gene for chromodomain-helicase-DNA binding protein 5 (CHD5), a chromatin-remodeling protein, as a bona fide suppressor gene in this region.90 Low expression of CHD5 is strongly correlated with unfavorable clinical features, biologic features, and outcome.90 Other potential tumor suppressor genes (miR34a, KIF1BB, CASZ1, ARID1A) have been identified in this region,72,91,92,93,94,95,96 but none are located in the minimal common region of deletion. Nevertheless, most 1p36 deletions are relatively large, so it is likely that more than one tumor suppressor gene may play a role in neuroblastoma pathogenesis.
Allelic loss of 11q is present in 25% of primary neuroblastomas.97,98,99 Interestingly, this genomic aberration is rarely seen in tumors with MYCN amplification, but is still highly associated with other high-risk features such as advanced stage, older age, and unfavorable pathology. Therefore, deletion of 11q is a strong prognostic marker of unfavorable disease in the subset of tumors without MYCN amplification.99,100 Although the tumor suppressor gene that is the presumptive target of 11q deletions is not certain, CADM1 at 11q23 has been proposed as a candidate.101 This gene encodes a transmembrane glycoprotein involved in cell-cell interactions and functional studies support a potential role in invasion and metastasis.
Allelic loss has been reported at multiple other chromosomal loci. Recent evidence suggests that 3p deletions often occur coincidentally with 11q deletions and help define a subset of patients with aggressive disease who lack MYCN amplification.99,100,102 Other genomic regions showing hemizygous deletions that have potential biologic relevance include 4p, 9p, 14q, and 19q.88,103,104 Although bona fide suppressor genes are being sought from each of these regions, such hemizygous alterations may preferentially target regions with numerous genes that negatively regulate cell survival, rather than targeting single genes for biallelic inactivation.105
Gene Expression and Molecular Signatures in Neuroblastomas
Molecular Signatures in Neuroblastoma
Just as improved genomic technologies have led to new insights into the DNA alterations occurring in neuroblastoma, robust gene expression platforms have led to insights into neuroblastoma biology. Transcriptome datasets have identified relevant biopathways associated with specific tumor subtypes.40,106,107,108,109 Examples include identification of polyamine metabolism as a tractable therapeutic target downstream of MYCN amplification,110,111 and enrichment in sympathoadrenal developmental genes in favorable, low-risk disease.112,113,114 Interestingly, independent datasets have implicated MYC signaling in neuroblastomas with poor outcome that do not have MYCN gene amplification,112,115,116 suggesting that deregulated MYC is essential to the high-risk phenotype. Molecular signatures have also been sought to improve current risk classification schemes, which currently rely on clinical features and genomic changes in the tumor. Numerous gene lists and classifiers have been shown to identify risk class for neuroblastomas at diagnosis,107,109,117,118 or to provide prognostic information for specific subsets, such as older children with metastatic MYCN-nonamplified disease.106 Their utility in patient management awaits robust validation in prospective clinical trials and will require efforts to increase the percentage of patients from whom high-quality tumor RNA can be obtained in a timely manner for these purposes.
Expression of RTKs
The neurotrophin receptors (NTRK1, NTRK2, and NTRK3 encoding TrkA, TrkB, and TrkC) and their ligands (NGF, BDNF, neurotrophin-3, respectively) are important regulators of survival, growth, and differentiation of neural cells.119 Their temporospatial expression plays a critical role in the normal development of the sympathetic nervous system, and their co-opted signaling correlates
with neuroblastoma phenotype. High TrkA expression correlates with younger age at diagnosis, lower tumor stage, favorable biologic features, and good outcome.43,44,45 It has been proposed that activation of TrkA by NGF present in the tumor microenvironment promotes differentiation into mature ganglion cells, whereas NGF deprivation initiates apoptosis, with TrkA functioning as a dependence receptor.44,120 Thus, the regression seen in TrkA-expressing neuroblastomas in infants may be due, at least in part, to delayed activation of developmentally programmed cell death resulting from NGF deprivation.
with neuroblastoma phenotype. High TrkA expression correlates with younger age at diagnosis, lower tumor stage, favorable biologic features, and good outcome.43,44,45 It has been proposed that activation of TrkA by NGF present in the tumor microenvironment promotes differentiation into mature ganglion cells, whereas NGF deprivation initiates apoptosis, with TrkA functioning as a dependence receptor.44,120 Thus, the regression seen in TrkA-expressing neuroblastomas in infants may be due, at least in part, to delayed activation of developmentally programmed cell death resulting from NGF deprivation.
In contrast to TrkA, expression of TrkB is strongly associated with aggressive tumor behavior and unfavorable biologic features, such as MYCN amplification.44 These tumors also commonly express the cognate TrkB ligand BDNF,121 which may represent an autocrine or paracrine survival pathway. Constitutive activation of the TrkB/BDNF pathway also contributes to chemotherapeutic drug resistance and metastasis, perhaps through suppression of anoikis.121,122,123,124 These characteristics make targeted inhibition of this pathway an attractive therapeutic goal. The independent role of TrkC expression remains to be elucidated, as TrkC expression occurs in a subset of tumors that also express TrkA.125,126 Another transmembrane neurotrophin receptor called p75 (p75NTR; TNFRSF16) binds all the NGF family of neurotrophins with low affinity,43,44,45 but its biologic and prognostic significance independent of Trk receptor expression is unclear.127 Several other RTKs involved in normal neuronal development have been implicated in NB biology, such as ALK (discussed above) RET, epidermal growth factor receptor (EGFR), and others.128,129 Although their role in neuroblastoma is still unclear, these RTKs represent potential therapeutic targets, at least for subsets of patients.
PATHOLOGY
Neuroblastoma is one of the “small, round blue-cell” neoplasms of childhood, which includes sarcomas, lymphomas, and others. However, an experienced pathologist usually has little difficulty discriminating neuroblastoma from these other neoplasms, particularly with immunohistochemical stains as well as molecular testing to complement conventional light microscopy. Neuroblastomas will stain with neural-specific antibodies recognizing TH, neuron-specific enolase, synaptophysin, chromogranin, and others. Neuroblastomas presumably arise from sympathoadrenal precursors, and the sites in which neuroblastomas arise correlate with normal tissues of the sympathetic nervous system, including the adrenal medulla.
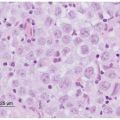
The three classic histopathologic patterns of neuroblastoma, ganglioneuroblastoma, and ganglioneuroma reflect a spectrum of neuronal differentiation (Fig. 30.4). The typical neuroblastoma is composed of small, uniformly sized cells containing dense, hyperchromatic nuclei and scant cytoplasm. The presence of neuritic processes, or neuropil, is a characteristic feature of all but the most primitive neuroblastomas. The Homer-Wright pseudorosette is a ring of neuroblasts surrounding a central core of eosinophilic neuropil. Although a classic finding in neuroblastomas, they are infrequently observed in primary or metastatic tumor tissue.
The fully differentiated, benign counterpart of neuroblastoma is a ganglioneuroma. It is composed of mature ganglion cells, neuropil, and Schwannian stroma. Ganglioneuroblastoma defines a heterogeneous group of tumors with histopathologic features spanning the extremes of maturation represented by neuroblastoma and ganglioneuroma. The term “maturing neuroblastoma” is used for tumors that contain less than 50% maturing or mature ganglion cells, and “ganglioneuroblastoma” for tumors with more extensive maturation. Ganglioneuroblastomas may be either nodular or diffuse, depending on the gross pattern seen, but diffuse ganglioneuroblastoma is associated with less aggressive behavior (Fig. 30.4). It is increasingly clear that focal ganglioneuroblastomas frequently behave aggressively,130,131 and the neuroblastic component may harbor genomic alterations such as MYCN amplification.59 Therefore, multiple sections, especially from regions with different gross appearance, should be examined to properly categorize tumors by histopathologic and genetic criteria.
Shimada et al.130 developed a prognostic classification for neuroblastoma based on histopathologic features, such as the presence or absence of Schwannian stroma, the degree of differentiation, and the mitotic-karrhyorectic index, to classify tumors as either “favorable” or “unfavorable.” Indeed, an international group of pediatric pathologists including Shimada modified this into the International Neuroblastoma Pathology Classification (INPC).132,133 This classification incorporates other features to improve concordance among pathologists implementing this system. However, because this system still utilized patient age, select histologic features have been incorporated into the International Neuroblastoma Risk Group (INRG) classification system.134 The INRG risk group classification system incorporates the histologic category and grade of tumor differentiation, without confounding the independent prognostic significance of these findings by incorporating age with histology.
CLINICAL PRESENTATION
Neuroblastomas can arise from the adrenal gland or any site along the sympathetic chain. The locations of primary tumors at the time of diagnosis are varied and change with age. Most primary tumors occur within the abdomen (75%), although the frequency of adrenal tumors is slightly higher in children (40%) compared with infants (25%).3 Infants also have more thoracic and cervical primary tumors. The majority of children with neuroblastoma are diagnosed by the age of 5 years, but the disease does occur in adolescents and occasionally adults (Fig. 30.1). Clinical behavior is more indolent in older patients.135,136
Neuroblastoma can metastasize by lymphatic and hematogenous dissemination. Regional lymph node metastases are found in up to 35% of patients with apparently localized tumors, and 30% of patients with metastatic disease also have regional lymph node involvement.137 Spread of tumor to lymph nodes outside the cavity of origin is considered to be metastatic disease, but these patients may have a better outlook if there is no other evidence of distant metastases.138 Hematogenous spread occurs most frequently to bone marrow, cortical bone, liver, and skin (subcutaneous tissue). Rarely, disease may spread to lungs or the central nervous system at diagnosis,139 but this is more commonly a manifestation of recurrent or end-stage disease. Neuroblastoma can disseminate to the central nervous system by inward compression on the brain from cranial metastases, by hematogenous spread or by meningeal involvement through the cerebral spinal fluid.140 The proportion of patients presenting with localized, regional, or metastatic disease is age dependent.141,142,143
The signs and symptoms of neuroblastoma reflect the location of primary, regional, and metastatic disease. Abdominal disease results in complaints of fullness or discomfort, but biologically favorable disease can present as an asymptomatic mass, or even be identified incidentally. Physical examination commonly reveals a fixed, hard abdominal mass; the borders may be difficult to define due to the retroperitoneal origin. If primary tumors arise from the organ of Zuckerkandl, bladder and bowel symptoms may occur as a result of direct compression. Massive involvement of the liver with metastatic disease is particularly frequent in infants and may result in respiratory compromise.
Primary thoracic tumors present as symptomatic masses or can be discovered incidentally. High thoracic and cervical masses can be associated with Horner syndrome (unilateral ptosis, myosis, anhydrosis) or rarely anisocoria. Occasionally, large thoracic tumors are associated with mechanical obstruction leading to superior vena cava syndrome. Paraspinal tumors in the thoracic, abdominal, and pelvic regions may extend into the neural foramina of the vertebral bodies and cause neurological symptoms from compression of nerve roots and spinal cord. The range of symptomatology
includes subacute or acute paraplegia, bladder or bowel dysfunction, or less commonly radicular pain. This situation can be a medical emergency (see Chapter 38), although the optimal approach to managing spinal cord compression is controversial (see below).
includes subacute or acute paraplegia, bladder or bowel dysfunction, or less commonly radicular pain. This situation can be a medical emergency (see Chapter 38), although the optimal approach to managing spinal cord compression is controversial (see below).
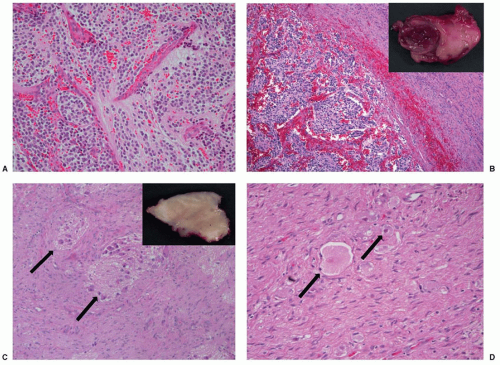 Figure 30.4 Pathologic appearances of neuroblastoma subtypes (all histologic preparations stained with hematoxylin and eosin). A: Poorly differentiated neuroblastoma, with nests of neuroblasts in a background of light pink fibrillary neuropil (magnification ×100). B: Nodular ganglioneuroblastoma, defined by the presence of a grossly visible hemorrhagic nodule (inset). Histologic examination reveals an expansile hemorrhagic nodule of neuroblastoma (lower left) in a background of Schwannian stroma (right side of the image) (magnification ×40). C: Intermixed ganglioneuroblastoma, with microscopic nests of neuroblastoma (arrows), embedded in Schwannian stroma (magnification ×100). The nests contain neuroblasts and fibrillary neuropil, which is less dense than the surrounding stroma. Grossly the tumor is tan, glistening, and uniform, with no gross nodularity (inset). D: Ganglioneuroma, composed of Schwannian stroma and scattered maturing ganglion cells (arrows). Immature neuroblasts and neuropil are absent (magnification ×200). (Courtesy of Dr. Bruce Pawel, The Children’s Hospital of Philadelphia and The University of Pennsylvania School of Medicine.) |
Several classical signs and symptoms have been associated with metastatic neuroblastoma. Proptosis and periorbital ecchymoses are frequent and result from tumor infiltration of periorbital bones, but the reason for the predilection remains obscure. Widespread bone and bone marrow disease causes bone pain, which can lead to limping, or irritability in a younger child. In addition, there may be symptoms of bone marrow replacement such as anemia, bleeding, or infection. Skin involvement is seen almost exclusively in infants and is characterized by nontender, bluish subcutaneous nodules.
Neuroblastoma in adolescents and adults is rare, as only about 2% of patients are over 10 years of age (Fig. 30.1). Although the distribution of primary sites is similar to that seen in younger children, the course of the disease is generally more indolent, and it is frequently fatal.136,144,145 MYCN amplification is rarely observed in these patients, but they have other genomic changes that are characteristic of unfavorable tumors, such as somatically acquired mutations in ATRX71 and ALK.136,144 There may be a role for radiation therapy in localized disease,136 but metastatic disease in these patients is rarely treated successfully with conventional therapy due to de novo or acquired chemoresistance.
Paraneoplastic Syndromes
Opsoclonus-myoclonus-ataxia syndrome (OMAS) is a unique paraneoplastic syndrome that is observed in 2% to 3% of newly diagnosed neuroblastoma patients.146,147,148,149 OMAS is manifested by rapid and chaotic eye movements, ataxia, and myoclonia. Most children with this syndrome have localized disease and a favorable tumor-related outcome, as this syndrome is correlated with an immune-mediated antitumor host response.148,150,151,152 However, 70% to 80% of these children have long-term neurologic deficits, including cognitive and motor delays, language deficits, and behavioral abnormalities.146,147,148,149 These are presumably due to antineural cells or antibodies directed against the tumor that cross-react with neural cells in the central nervous system.148,150,151,152 Tumor removal may result in at least temporary improvement of neurologic symptomatology, but recurrence or persistence of OMAS symptoms is common, and exacerbations are often associated with intercurrent infections. About 30% to 50% of OMAS cases occur without associated neuroblastoma,153 although some of these cases may be due to tumors that spontaneously regressed. All children with OMAS should have a complete diagnostic workup for neuroblastoma including scintigraphy with 123I-meta-iodo-benzylguanidine (123I-MIBG).154 However, there is now consensus that immunosuppressive therapy is indicated to treat OMAS. Ongoing clinical trials are exploring the role of intravenous immunoglobulin,
chemotherapy, glucocorticoids, rituximab, and/or other agents in managing this syndrome.155,156,157
chemotherapy, glucocorticoids, rituximab, and/or other agents in managing this syndrome.155,156,157
Vasoactive intestinal peptide (VIP) syndrome consists of intractable watery diarrhea associated with abdominal distension, hypokalemia, and dehydration.158,159,160 Most tumors associated with this syndrome secrete VIP and are mature histologically (ganglioneuroblastoma or ganglioneuroma), and these patients almost always have a favorable tumor-related outcome.161 Unlike OMAS, surgical removal of the tumor usually results in complete resolution of symptoms, because tumor secretion of VIP is responsible for the syndrome.
CLINICAL AND LABORATORY EVALUATION
Physical examination should include detailed evaluation for an abdominal mass and estimation of location and size. Liver size should also be evaluated. Detailed evaluation of all lymph node structures should be performed, with note made of any enlarged, discolored, nontender nodes. An enlarged left supraclavicular node may be seen in high-risk neuroblastoma patients who have intra-abdominal disease with nodal spread. Detailed head-and-neck evaluation for proptosis, signs of Horner syndrome, anisocoria, or skull-based metastases should be performed. Detailed neurologic examination is required, especially in young patients with paraspinal masses, to ensure that spinal cord impingement is not missed.
To confirm a diagnosis of neuroblastoma, histologic evidence is required that demonstrates neural origin or differentiation by light microscopy or immunohistochemistry. Alternatively, because the bone marrow is frequently involved, some patients may be diagnosed with neuroblastoma on the basis of the presence of “unequivocal” neuroblastic cells involving the bone marrow, accompanied by increased urinary catecholamine metabolites. Because tumor-specific genomic markers and histopathologic evaluation are critical determinants for diagnosis, risk assessment, and treatment planning, especially for younger children, sufficient tumor-containing material should be obtained if possible. An involved marrow specimen may be adequate for genomic studies in some cases, but a primary tumor biopsy should be performed for treatment planning if adequate tumor tissue cannot be obtained by other means.
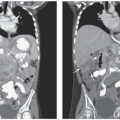
The goal of diagnostic testing is to definitively establish the diagnosis and precisely define the extent of disease. MRI or CT are the preferred methods for evaluation of the primary tumor in the neck, chest, abdomen, and pelvis (Fig. 30.5). MRI evaluation may be superior for paraspinal lesions and is essential when evaluating intraforaminal extension with the potential for cord compression. Indeed, MRI is replacing CT in many centers because of higher resolution and the absence of radiation exposure. Patients should also have evaluation for metastatic lesions, including an assessment of the bone marrow and bony skeleton. Exclusion of some of the tests recommended below may be considered in some settings (e.g., perinatal detection of a small adrenal mass), but only in a setting in which careful follow-up is ensured.
Evaluation of the bone marrow compartment. Patients should have bilateral bone marrow aspirates and trephine biopsies.162,163 Aspirate amounts should be sufficient for both standard histology and histochemical stains, as well as genomic testing if a tumor biopsy is not possible. Although light microscopy is generally considered sensitive to the level of 1 neuroblastic cell per 100 nucleated cells, immunohistochemical staining with neural-specific antibodies increases the sensitivity to at least 1 in 100,000 cells.164,165
Evaluation of the bony skeleton. 123I-MIBG scintigraphy has superior sensitivity and specificity over technetium bone scan or 131I-MIBG scintigraphy and has become an integral part of disease staging and response evaluation, especially in high-risk patients (Fig. 30.6).135,166 If 123I-MIBG scan is negative and bone metastases are strongly suspected, 99mTc-diphosphonate scintigraphy bone scan, plain radiographies, or MRI may be useful, depending on the age of the patient. Positron emission tomography with 18F-deoxyglucose is a useful metastatic disease evaluation but may be less sensitive than MIBG scintigraphy.167,168
The exact type of procedure used for diagnosis in a patient with suspected neuroblastoma who has an unresectable primary tumor may depend on the individual circumstances. The clinician must weigh the value of having adequate tissues for diagnosis and assessment of prognostic factors, while being cognizant of avoiding surgical complications. In the United States, most investigators have recommended limited open biopsy because it is easier to ensure hemostasis, the biopsy specimens are larger, and histopathology can still be assessed.132,133 However, some centers have experience using CT-guided needle biopsies and have obtained adequate materials for molecular studies.169 Although the care of the patient is paramount, most investigators also recognize the critical importance of submitting residual biologic materials (beyond those required for diagnosis) to tumor banks that support translational research.
Catecholamine Metabolism
Because of their noradrenergic derivation, neuroblastomas typically express essential enzymes involved in catecholamine synthesis, affording a technique for noninvasive detection of tumor markers. A simplified diagram of catecholamine synthesis and metabolism is depicted in Figure. 30.7. Because monamine oxidase and catechol-O-methyltransferase enzymes are typically expressed in neuroblastomas, HVA and VMA are the metabolites that show the highest sensitivity and specificity for tumor detection. Using high-pressure liquid chromatography technology, the sensitivity and specificity can approach 100% without significant influence from diet other confounding factors.170 Screening for HVA and VMA in urine samples (normalized per mg creatinine) should be part of every diagnostic workup if neuroblastoma is in the differential diagnosis. These markers also provide a relatively reliable method for monitoring response to therapy and screening for occult relapse after the completion of therapy.
Diagnostic Criteria
On the basis of international consensus,162,163 a diagnosis of neuroblastoma is established: (a) if unequivocal pathologic diagnosis is made from tumor tissue by light microscopy, with or without immunohistology; or (b) if a bone marrow aspirate or trephine biopsy contains unequivocal tumor cells (e.g., syncytia or immunocytologically positive clumps of cells) and there are increased urinary catecholamine metabolites. Tumor-specific biologic information is particularly important for risk stratification of infants, so adequate tumor tissue should be obtained if at all possible. Because of data suggesting a later age cutoff for risk classification,142,171,172 it may be warranted to encourage tumor biopsy for children younger than 18 months of age to ensure acquisition of adequate tumor material for histology and molecular studies.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


