All MPNs have one or more shared features:
1.
Overproduction of one or more blood cell lines, i.e., platelets, red blood cells, and mature myeloid cells.
2.
Hypercellular/fibrotic marrow
3.
Cytogenetic abnormalities.
4.
Extramedullary hematopoiesis
5.
Transformation to acute leukemia
6.
Overlapping clinical features
2 Mutations in Philadelphia-Negative MPNs (PV, ET, MF)
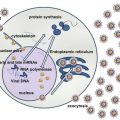
JAK2, MPL, and CALR are the three major mutations in MPNs occurring in 99 % of patients with PV and 85 % in ET and MF. JAK-STAT pathway is activated in all MPNs regardless of the founding driver mutations [3]. JAK2 belongs to non-receptor tyrosine kinase family and plays a fundamental role in hematopoiesis. Normal JAK2 activation requires ligand binding for signal transduction through the STATS transcriptional pathway. In case of JAK2 mutation ligand binding is not required, continuous down stream signaling occurs. The aberrant activation of the signal transduction pathways results in increased proliferation and inhibition of the apoptotic pathways of the JAK2 mutant myeloid stem cells together with hypersensitivity to several cytokines including erythropoietin, GM-CSF, IL-3, and insulin-like growth factor-1 (IGF-1).
Overall prevalence of JAK2V617F mutations in MPNs is 75 %, with 95 % mutations occurring in PV [4]. Phenotypic differentiation occurs due to differences in host genetic modifications, allele burden, gender, loss of heterozygosity, and qualitative and quantitative differences in downstream signaling pathways. The clones of JAK2V617F-mutated stem cells remain stable over prolonged periods accounting for the indolent nature and clinical stability of PV and ET. MPL10 exon mutations located on the thrombopoietin receptor is present in 5 % ET and PMF.
Somatic mutation of the CALR gene encoding calretinin occurs in ET/MF patients with unmutated JAK2/MPL. Normal functions of CALR include normal folding of newly synthesized glycoproteins, calcium hemostasis, immunogenic cell death, proliferation, and apoptosis. Diverse epigenetic mutations involving other metabolic pathways are acquired during the course of disease, which include DNA methylation genes TET2, DNMT3A, and IDH1/2 and chromatin structure genes EZH2 and ASXLI for evolution of ET/PV into AML [5].
There is NO indication of monitoring bone marrow (BM) response or determining sequential assessment of JAK2V617F allele burden. BM aspirate and biopsy are indicated only if disease progression and transformation to MF/AML are suspected.
Goals of Therapy for PV/ET
1.
To avoid occurrence/recurrence of thrombosis or bleeding
2.
To minimize/delay disease progression to post ET-MF or acute leukemia
3.
To control systemic symptoms
4.
To treat complications of hemorrhage/thrombosis
5.
To manage special risk situations, e.g., Pregnancy and surgery
3 Polycythemia Vera (PV)
PV is a chronic disorder with increased blood viscosity and sluggish blood flow due to increased red cell production. Median survival is 14.1 years, risk of late progression to PV-MF is 15–20 %, and development to acute leukemia is 3–5 % [5]. The prognosis of patients with PV-acute leukemia is dismal with a median survival of 2–3 months [6].
In peripheral blood there is an increase in all three-cell lines. Thrombocytosis may contribute to bleeding and thrombosis. Platelet aggregation abnormalities are common but do not correlate with bleeding and thrombotic activities.
The bone marrow shows panmyelosis, with increase in the erythroid compartment and to a lesser extent of the myeloid and the megakaryocyte series. The red cell mass is increased and it is extremely sensitive to cytokines such as erythropoietin, IL-3, GM-CSF, and steel factor.
3.1 Criteria for Diagnosis of PV
Proposed WHO diagnostic criteria for PV for 2014 are listed in Tables 2 [7]. These include 3 major and 1 minor criterion.
Table 2
2014 proposed revision for World Health Organization (WHO) diagnostic criteria for BCR-ABL negative myeloproliferative neoplasms
Polycythemia vera (PV)a | Essential thrombocythemia (ET)b | Primary myelofibrosis (PMF)c |
|---|---|---|
Major criteria | Major criteria | Major criteria |
1. Hemoglobin > 16.5 g/dl (men) > 16 g/dl (women) or hematrocit >49 % (men) > 48 % (women) | Platelet count ≥450 109/l | Megakaryocyte proliferation and atypia, |
2. BM trilineage myeloproliferation with pleomorphic megakaryocytes | Megakaryocyte proliferation with large and mature morphology | Accompanied by either reticuline and/or collagen |
3. Presence of JAK2 mutations | Not meeting WHO criteria for CML, PV, PMF, MDS, or other myeloid neoplasm | Fibrosis |
Presence of JAK2, CALR, or MPL mutation | Not meeting WHO criteria for CML, PV, ET, MDS, or other myeloid neoplasms | |
Megakaryocyte proliferation with large and mature morphology | Presence of JAK2, CALR, or MPL mutation | |
Minor criteria | Minor Criteria | Minor criteria |
1. Subnormal serum erythropoietin level | Presence of a clonal marker (e.g., abnormal karyotype) or absence of evidence for reactive thrombocytosis | Presence of a clonal markers (e.g., abnormal karyotype) or absence of evidence for reactive bone marrow fibrosis |
Presence of anemia or palpable splenomegaly | ||
Presence of leukoerythroblastosis, or increased lactate dehydrogenasef | ||
Requirement for Diagnosis | Requirement for Diagnosis | Requirement for Diagnosis |
Meeting either all 3 major criteria or the first 2 major criteria and 1 minor criterion | Meeting all 4 major criteria or first 3 major criteria and 1 minor criterion | Meeting all 3 majors criteria or the first 2 major criteria and all 3 minor criteria |
Secondary polycythemia should be ruled out in patients with isolated erythrocytosis. These include smoking, pulmonary or cardiac problems, overweight with nocturnal dyspnea, hepatic or renal tumors, or hemoglobinopathies.
3.2 PV Signs and Symptoms
In developed countries, patients are frequently diagnosed during a routine peripheral blood smear or for investigations of vague symptoms: unexplained weakness and pruritus (after taking a shower with warm water and physical exercise and at bed time or when changing clothes). Patients in developing countries often present with symptoms of hyperviscosity. These include headaches, blurring of vision, plethora, arthralgia, abdominal discomfort, excess sweating erythromelalgia, and cyanosis. Ulceration and gangrene occur due to thrombosis in small blood vessels. Gout, renal stones, and hyperuricemia are due to high red cell turnover. Mild gingival or GI bleeding, stomach ulcers, epistaxis, and bruising may occur occasionally. Splenomegaly is present in 30–40 % patients. Financial constraints, lack of health insurances, and access to medical care are major limitations face by patients in our part of the world from presenting early in the course of disease.
Thrombotic events: The frequency of venous and arterial venous thrombotic events is directly proportional to the rise in hematocrit. Cerebral blood flow also reduces when hematocrit goes above 53 %. Major thrombotic events in PV range from 34 to 39 %; hemorrhagic events are somewhat less common. Both contribute to increased morbidity and mortality in the patients. More severe thrombotic events include myocardial infarction, stroke, and TIA. Rare thrombotic events include splanchnic vein thrombosis (SVC): splenic, hepatic, portal, and mesenteric vein thrombosis and Budd-Chiari syndrome (BCS) [8]. CT/US scans help in determining asymptomatic splanchnic vein thrombosis.
3.3 Treatment for PV
The cornerstone for treatment for PV is to normalize blood counts and to maintain a hematocrit of <45 % in order to decrease blood viscosity and reduce thrombotic episodes. Available therapies do not prevent disease progression to PV-MF or acute leukemia. Patients are stratified according to the risk of thrombosis and not based on survival or disease progression. Prognostic model for PV based on (1) age, (2) prior venous thrombosis is used to stratify patients into low risk (age < 60 years) and high risk (age >60 years) and/or thrombosis [4].
Correction of cardiovascular risk factors: Lifestyle risk factors need to be modified in all patients, so they are not contributory to the already increased risk of vascular complications in PV. These include weight reduction, cessation of smoking, physical exercise, normalizing lipid profile, and avoiding oral contraceptives and situations which increase risk of thrombosis.
Antiplatelet therapy: Low-dose aspirin is recommended to all patients except those with acquired von Willebrand disease, extremely high platelet counts, and peptic ulcer disease. The ECLAP trial randomized patients with PV to low-dose aspirin 100 mg/daily versus placebo. There was a significant reduction in nonfatal myocardial infarction, nonfatal stroke, pulmonary embolism, major venous thrombosis, or death from cardiovascular complications (RR = 0.40, CI 0.18-0.91; p = 0.03) [4, 9].
Low-risk patients: Low-risk patients are treated with phlebotomy and aspirin alone to maintain a hematocrit of <45 %. Rates of cardiovascular deaths and major thrombosis [4, 9] are significantly reduced. Systemic symptoms like pruritus and those due to enlarged spleen cannot be controlled by phlebotomy alone. In younger patients phlebotomy is tolerated better and 350–400 mL of blood can be removed safely. Smaller volumes of blood should be removed in older patients.
High–risk patients: Cytoreductive therapy is indicated in patients who present initially with high-risk disease, symptomatic splenomegaly, extremely high platelet counts, and leukocytosis of >20,000 × 109/L or those who progress from low-risk to high-risk group.
Therapeutic options for cytoreduction in high-risk patients include HU, interferon alpha, anagrelide, alkylating agents, busulfan, and JAK2 inhibitors which have recently been approved in the USA [10].
Hydroxyurea (HU) is a non-leukemogenic oral antimetabolite that inhibits DNA synthesis and repair. It is the initial treatment of choice for patients requiring cytoreductive therapy. It is cheap, is well tolerated, and effectively reduces elevated counts, spleen size, and thrombotic risks. Initial dose starts from 15 to 20 mg/kg until response to therapy followed by maintenance [11]. Side effects of HU include myelosuppression, macrocytosis, leg and mouth ulcers, raised creatinine, and skin and nail changes.
Interferon/PEG-IFN although not approved are used in patients intolerant or resistant to HU, in patients who are pregnant, and in patients with intractable pruritus. Complete hematologic responses occur in approximately 76 % patients with PV. The mutated JAK2 allele burden is also decreased. In 15–20 % patients IFNs cause complete disappearance of the mutant clone with durable complete remissions [12]. Serious adverse effects include increased risk of depression, hypothyroidism, and retinitis and exacerbation of autoimmune disorders, neuritis, and fatigue.
Based on the RESPONSE trial JAK2 inhibitor ruxolitinib is currently used in patients intolerant/resistant to HU [13]. Precise mechanism of action is unknown, but there is inhibition of JAK2 mutant clones and downregulation of cytokines, but no complete eradication of the malignant clones. Response is not dependent on JAK2 mutational status. Complete hematological response was seen in 24% of patients, symptoms improvement occured in 50% of the patients, 35% of patients had reduction in spleen volume. Thromboembolic events occured lower frequency compared tp patients on hydroxyurea (2.8 events per 100 patients per year).Fewer patients required phlebotomies. Overall Ruxolitinib is well tolerated.
3.3.1 Special Situations
Thrombosis: For splanchnic vein thrombosis, low-molecular-weight heparin (LMWH) followed by lifelong anticoagulation is also indicated [14].
Surgery: Thrombosis or hemorrhage occurs in approximately 8 % of patients with PV/ET after surgery [15]. Prophylactic LMWH and normalization of hematocrit are important for prevention.
Pregnancy: It is uncommon but requires special care. In low-risk patients with no history of abortions, aspirin is given during pregnancy along with 6 weeks of LMWH after delivery. Hematocrit is maintained at < 40 %. For women with pregnancy complications and/or thrombosis, LMWH is used throughout pregnancy and control of hematocrit and thrombocytosis is done through IFN [16].
Pruritus: It may become very debilitating; antihistamines are usually ineffective. IFN, ruxolitinib, and in occasional cases UV light and psoralen are effective.
4 Essential Thrombocythemia (ET)
There is sustained proliferation of megakaryocyte progenitors and increased platelet production, due to extreme susceptibility to cytokines including IL-6, IL-3, and thrombopoietin [17]. Persistent platelets counts > 450 × 109/L is the threshold for diagnosis of possible ET. Higher platelet counts are associated with hemorrhagic and thrombotic complications due to acquired von Willebrand disease with abnormalities in platelet aggregation studies [18]. Lowering of platelets counts is associated with correction of defects and cessation of bleeding disorders.
The JAK2V617F occurs in approximately 50–60 % patients with ET. CALR mutation is found in 60–88 % patients with ET with negative JAK2 mutation. CALR-mutated patients with ET are predominantly males, have younger age, have higher platelet count, and have lower WBC counts and hemoglobin with a lower incidence of thrombosis as compared to JAK2-mutated patients [19].
4.1 Diagnosis
The 2014 WHO proposed major diagnostic criteria for ET are listed in Table 1
Causes for reactive thrombocytosis have to be excluded, which include iron deficiency anemia, inflammation, infections, malignancies, splenectomy, and cryoglobulinemia. Hemorrhagic or bleeding disorders have to be ruled out.
4.2 Signs and Symptoms
Asymptomatic patients may present with persistently elevated platelet counts. In symptomatic patients clinical signs and symptoms are similar to those with PV. Hemorrhagic or thrombotic episodes occur in both major blood vessels and microvasculature in 20–80 % of patients [17]. Thrombotic episodes occur more frequently in older people due to coexistence of microvascular disease. Pregnancy is associated with a higher incidence of spontaneous abortions due to placental thrombosis.
4.3 Treatment Options
ET is stratified into 3 risk groups.
1.
Low-risk group includes asymptomatic patients younger than 60 years with platelet counts of <1,500 × 109/L with no history of thrombosis, bleeding, or microvascular disease.
2.
High-risk group includes patients older than 60 years with history of thrombosis/bleeding or microvascular disease.
3.




Intermediate group includes all patients who cannot be placed in the above risk categories.
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
