Natural Products: Bleomycin and Trabectedin
Natural product research has yielded many active anticancer compounds acting as inhibitors of topoisomerases (Chapter 4) and as antimitotics (Chapter 4). Others, such as bleomycin and trabectedin, interact directly with DNA and exhibit unusual mechanisms of action.
BLEOMYCIN
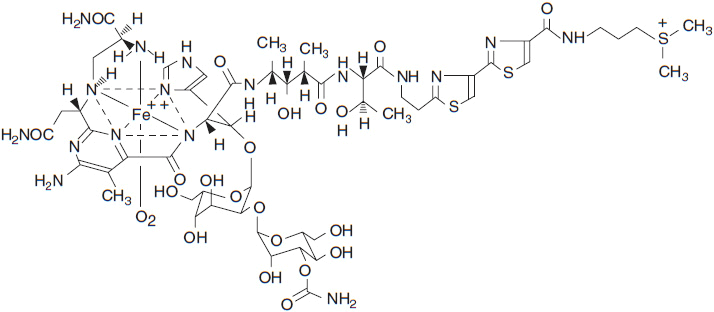
Traditional fermentation research has been the backbone of efforts to discover anti-infective and antitumor agents. From this research has come bleomycin, a drug important in the curative combination therapy of Hodgkin disease and testicular germ cell tumors. Among the exquisite cytotoxics designed by nature, the most unique is bleomycin, which was isolated from the broth of the fungus, Streptomyces verticillus. The clinical bleomycin preparation consists of a family of peptides that share a common C-terminal metal binding core, attached to a variable DNA binding portion with different substitutions on the amino-terminal end of the molecule (Figure 7-1). The various bleomycin peptides have a molecular weight of about 1500, and differ in their potency for DNA cleavage, but share a common chemistry. The predominant peptide, bleomycin A2, comprises 70% of the clinical preparation.
 MECHANISM OF ACTION
MECHANISM OF ACTION
The bleomycin peptides function as carriers for a catalytic metal that undergoes rapid cycles of oxidation/reduction (1). The metal may be copper, iron, cobalt, zinc, or others. The metal ligand is bound in a coordination complex of amine groups contributed by the unusual amino acids of the peptide. All metals but the cobalt species are readily exchangeable; thus 57-Co (II) bleomycin is sufficiently stable to be used as a tumor-imaging agent. The clinical preparation of bleomycin is metal free, but upon administration rapidly acquires Cu (II) or Fe (II), the latter probably representing the active form of the drug. Bleomycin binds to DNA by intercalation of its C-terminal bithiazole groups between guanine bases in adjacent DNA strands. Intercalation brings the bleomycin metal group into proximity with the deoxyribose backbone of DNA. The radicals released by cycles of oxidation/reduction of the metal core oxidize adjacent deoxyribose groups at the 3′-4′ bond, releasing propenal or propenal-thymine as its most frequent products (Figure 7-2). DNA single and double strand breaks (in the ratio of 10:1) result and must be repaired if the cell is to survive. If double strand breaks are sufficient in number, the cell undergoes apoptosis. Cells deficient in double strand break repair, such as those from tumors with BRCA1 mutation, are highly sensitive to bleomycin (2).
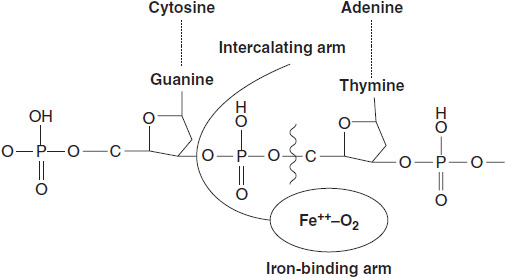
FIGURE 7-2 Intercalation of bithiazole groups between DNA strands in which one strand has the preferred sequence of GpT or GpC. The Fe++–O2 complex bound to bleomycin comes in opposition to the deoxyribose of DNA, abstracting a proton and cleaving the deoxyribose phosphate bond.
Bleomycin, a large, positively charged molecule, crosses the cell membrane slowly, possibly entering through vesicles. In the cytoplasm it is subject to cleavage by a specific aminohydrolase (AH), found in both malignant and normal tissues. AH may play a role in determining both response and toxicity (3). AH is found in low concentrations in lung tissue and skin, both of which are highly susceptible to bleomycin toxicity. Mice deficient in AH are extremely susceptible to the drug’s toxicity, while tumor cells with high levels of AH are resistant to the drug.
Other factors contribute to bleomycin sensitivity in experimental studies. Defective double strand break repair leads to hypersensitivity to the drug (3). Resistant cell may have increased capacity for detoxification of free radicals (4).
 CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
Bleomycin is administered i.v. or i.m. in doses of 10–20 units/m2 every 2 weeks. Higher doses are associated with an increased risk of pulmonary toxicity. The disposition of bleomycin is characterized by its slow uptake in tissues and selective hydrolysis in certain tissues, including liver and kidney. Most of an administered dose is excreted in the urine as an intact molecule. After intramuscular or intravenous administration, it disappears from plasma with a half-life of 2–3 h. Patients with renal dysfunction exhibit much delayed clearance and may develop overwhelming systemic toxicity (see below). Doses should be reduced at least 50% in patients with moderately reduced renal function (creatinine clearance 30–80 ml/min), and bleomycin should not be given to patients in severe renal failure (creatinine clearance below 30 ml/min) (5).
Bleomycin is also instilled intrapleurally (60 mg or greater, dissolved in 50–100 ml of saline) to ablate the pleural space. Given in this way it has a 75% success rate for ablation of effusions and has little systemic toxicity (6).
 TOXICITY
TOXICITY
The primary acute toxicity of bleomycin is cutaneous erythema, tenderness, and ulceration over the joints or the distal extremities. It may cause frank Raynaud’s phenomenon in occasional patients. Hyperpigmentation, nail changes, and alopecia may occur after prolonged use.
The most serious long-term toxic effects damage the lung. Pulmonary toxicity is dose related and most frequent (10% or greater) in patients receiving total doses of 450 mg or more, but may appear at lower total doses in selected patients, especially in those over 70 years of age, in those treated with single doses above 20 mg/m2, in those who are receiving chest irradiation or other pulmonary toxins such as gemcitabine (7) or the armed monoclonal antibody, SGN-35 (brevituximab). Pulmonary function may deteriorate acutely in patients exposed to high concentrations of inspired O2, as during surgery, subsequent to bleomycin treatment. In patients undergoing surgical resection of residual tumor after chemotherapy for germ cell tumors of the testis, oxygen must be used sparingly. In patients receiving a full course of therapy for Hodgkin disease, 20% have a greater than 20% decrease in diffuse capacity but most remain asymptomatic (8).
The pathophysiology of bleomycin pulmonary toxicity is not well understood. Experimental intertracheal injection of small doses of bleomycin or its terminal amine fragment evokes an acute inflammatory response and subsequent pulmonary fibrosis in mice and hamsters. Bleomycin induces macrophage and endothelial secretion of pro-inflammatory cytokines such as interleukins 1 and 6, TNF-alpha, and TGF-beta, and toxic lipid products such as phosphatidic acid. Mice deficient in metalloproteinases or plasminogen activators are resistant to bleomycin pulmonary fibrosis, suggesting that inhibitors of these enzymes might be useful in preventing fibrosis. A number of other experimental treatments block or ameliorate pulmonary toxicity of bleomycin, but as yet there are no clinical antidotes (9).
Bleomycin pulmonary toxicity initially manifests as a dry cough and shortness of breath, with or without fever. Alveolar infiltrates are seen on chest x-ray. Computerized tomography may reveal extensive infiltrates, even in patients with minimal evidence on routine chest x-rays, as well as pleural thickening and even cavitation. In later stages, there is evidence of extensive pulmonary parenchymal fibrosis. The acute syndrome must be distinguished from opportunistic infection or other causes of pulmonary infiltrates in cancer patients. In such patients, biopsy discloses inflammatory alveolar infiltrates, hyaline membrane formation, and fibrosis. Patients on bleomycin experience a steady decrease in pulmonary diffusion capacity with increasing total dose of drug, and, in later stages, develop evidence of restrictive disease and desaturation. There is no proven therapy for bleomycin pulmonary toxicity other than discontinuation of the drug. Glucocorticoids may produce temporary improvement of symptoms, but there is no evidence that glucocorticoids prevent or reverse pulmonary fibrosis. With discontinuation of the drug, the inflammatory component may resolve and pulmonary function may improve. Approximately a quarter of patients who develop extensive bleomycin toxicity, with symptoms, hypoxia, and bilateral infiltrates, will have a fatal outcome.
Limitation of total doses to less than 250 units, selection of patients without risk factors (see above), and careful clinical monitoring for evidence of toxicity have reduced the incidence of serious pulmonary injury to less than 5% in patients treated for Hodgkin disease or testicular cancer. The drug remains a unique and important component of their treatment.
TRABECTEDIN
Trabectedin belongs to a class of highly toxic natural products that bind to the minor groove of DNA and alkylate guanine at the N-2 position (Figure 7-3). Its DNA binding stabilizes double-stranded DNA, and stalls replication and transcription. It was isolated from the marine tunicate Ecteinacscidia turbinate and has a multi-ring structure with an isoquinoline group that interacts with transcription factors (TFs) and with the XPG component of nucleotide excision repair (NER). This interaction may contribute to its activity against NER proficient cells (10). In Europe, it is approved for second-line treatment of soft tissue sarcomas and, with Doxil, for treatment of ovarian cancer, and is available for compassionate use in the United States. A more potent and potentially less toxic analogue, xalypsis, which lacks the side chain and does not interact with NER (11), is in clinical development and appears to have lesser hepatic toxicity in early trials.
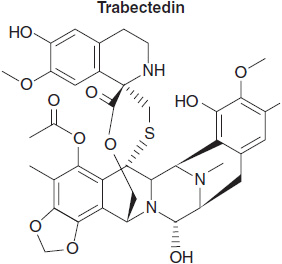
FIGURE 7-3 Trabectedin.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


