Phase
Year initiated
Description
Cancer indication
ClinicalTrials.gov identifier
0
2014
Silica imaging nanoparticle
Head and neck, melanoma, prostate, cervical, uterine
NCT02106598
0
2014
Magnetic nanoparticle thermoablation
Prostate
NCT02033447
0
2013
Iron oxide imaging nanoparticle
Head and neck
NCT01895829
1
2012
Anti-EGFR immunoliposome
Solid tumors
NCT01702129
1
2009
siRNA-loaded liposome
Solid tumors
NCT00882180
1
2011
siRNA-loaded liposome
Colorectal, pancreatic, gastric, breast, ovarian with hepatic metastases
NCT01437007
1
2009
siRNA-loaded liposome
Advanced solid tumors
NCT00938574
1
2012
siRNA-loaded liposome
Advanced cancers
NCT01591356
1
2014
siRNA-loaded liposome
Solid tumors, multiple myeloma, non-Hodgkins lymphoma
NCT02110563
1
2008
Iron oxide imaging nanoparticle
Brain neoplasms
NCT00769093
1
2014
Paclitaxel-loaded polymer micelle
Breast
NCT02064829
1
2011
Prostate-targeted docetaxel-loaded polymeric nanoparticle
Metastatic, solid tumors
NCT01300533
1
2008
Albumin-bound paclitaxel nanoparticle
Head and neck
NCT00736619
1
2007
Chemotherapeutic-polymer conjugate
Small cell lung, non-small cell lung
NCT00455052
1
2011
Magnetic biopsy needle and nanoparticles
Leukemia
NCT01411904
1
2011
Hafnium nanoparticles for targeted radiation
Adult soft tissue sarcoma
NCT01433068
1
2013
Hafnium nanoparticles for targeted radiation
Head and neck
NCT01946867
2
1999
Liposomal cisplatin
Ovarian
NCT00004083
2
2010
siRNA-loaded liposome
Neuroendocrine, adrenocortical
NCT01262235
2
2013
siRNA-loaded liposome
Pancreatic
NCT01808638
2
2011
Paclitaxel-loaded polymer micelle
Bladder, ureter
NCT01426126
2
2004
Albumin-bound paclitaxel nanoparticle
Breast
NCT00093145
2
2008
Chemotherapeutic-polymer conjugate
Breast
NCT00802945
2
2006
Chemotherapeutic-polymer conjugate
Solid tumors
NCT00333502
2
2011
Chemotherapeutic-polymer conjugate
Non-small cell lung
NCT01380769
2
2001
Chemotherapeutic-polymer conjugate
Fallopian tube, ovarian, primary peritoneal cavity
NCT00017017
2
2011
Chemotherapeutic-polymer conjugate
Glioblastoma multiforme
NCT01402063
2
2013
Prostate-targeted docetaxel-loaded polymeric nanoparticle
Prostate
NCT01812746
3
2012
Paclitaxel-loaded polymer micelle
Breast
NCT01644890
3
2008
Albumin-bound paclitaxel nanoparticle
Breast
NCT00785291
3
2007
Chemotherapeutic-polymer conjugate
Non-small cell lung
NCT00576225
3
2011
Chemotherapeutic-polymer conjugate
Breast
NCT01492101
4
2008
Liposomal paclitaxel
Solid tumors
NCT00606515
4
2009
Iron oxide imaging nanoparticle
Pancreatic
NCT00920023
2 Intro to Clinical Trials
The FDA as we know it today was born out of a 1937 tragedy in which 107 people died after taking the marketed “elixir” drug sulfanilamide, which prompted the government to enact more strict codes for food and drugs that could be legally sold in the United States [4]. Since then, the FDA approval process has continued to evolve in response to changes in drug technology, particularly in the realm of cancer therapy. Currently, there are three mandatory clinical trial phases through which a drug must go to be marketed in the United States, as well as a Phase 0 and a Phase IV which are sometimes required (Fig. 1). Given the lengthy, somewhat complex nature of clinical trials, as well as the wealth of cancer-treating nanoparticles spanning multiple phases, knowledge of the FDA approval process can provide a helpful context for evaluating the progress of a given treatment.
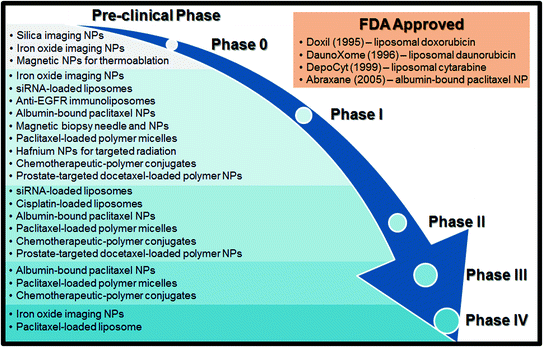
Fig. 1
A schematic of the FDA approval process with a list of clinical trials discussed in this book chapter
Before a therapy can reach clinical trials, it must be discovered, created, and tested both in vitro and in vivo. This period of invention and development is referred to as the “preclinical” phase and on average takes 6.5 years [5]. The ultimate goal of the preclinical period is to receive approval for an Investigational New Drug (IND) Application from the FDA. A successful IND lists the drug’s manufacturing information, clinical protocols, investigator information, and must also show a wealth of animal data suggesting positive pharmacological effects and tolerable toxicity [4]. In short, the transition from preclinical work to clinical trials requires a novel therapeutic that shows sufficient promise to be effective and safe in treating human disease. However, due to the diversity of nano-platforms, as well as the low number of nanoparticle therapeutics that have made it all the way to clinical approval, determining what constitutes a “promising” nanoparticle drug can be difficult; to this end, the National Cancer Institute developed the Nanotechnology Characterization Laboratory (NCL) in 2004 to address the standards for preclinical nanoparticle data [5, 6]. Researchers may apply via the NCL website (http://ncl.cancer.gov) to have their preclinical nanomaterials and devices extensively characterized in an “assay cascade,” that is, a battery of tests to determine physical properties, toxicology, pharmacology, efficacy, and many other parameters. The NCL has the infrastructure to perform this crucial preclinical testing on a large scale, allowing researchers to characterize their nanotechnology far more rigorously than they could alone. Moreover, this process can provide an accurate prediction of whether or not a technology will likely face significant efficacy or safety issues when it reaches clinical trials.
While many therapeutics will move directly from a successful IND to Phase I trials, some will first go through Phase 0 trials. Phase 0 trials, also developed by the National Cancer Institute, generally involve microdosing (dosing at sub therapeutic levels) of the drug in order to obtain relevant pharmacokinetic data; this process was developed largely as a means of streamlining the FDA approval process by serving as an early benchmark of the translatability of an IND [7]. As with the NCL, Phase 0 trials provide more clarity in the matter of which therapeutics are ready to take the leap from preclinical studies to the arduous FDA clinical trials.
The bulk of the FDA approval process, both in terms of time and capital, lies in Phases I–III. Phase I evaluates the safety of the new drug as its primary endpoint. Generally, 20–100 volunteers are enrolled, and the drug dose administered is gradually increased in order to observe patient response. These volunteers are usually healthy, though exceptions can be made for especially life-threatening illnesses [4]. Phase I generally lasts anywhere from 6 to 18 months, and about 2/3 of all INDs will progress to Phase II [5]. Phase II trials look at the efficacy of the drug. 100–300 patients are typically enrolled and the effective dose method of delivery, optimal dosing interval, and further safety concerns are all evaluated [4]. Phase II can last anywhere from 6 months to 2 years, and is typically where the majority of drugs fail, given that it is the first stage to really evaluate the effectiveness of the new drug in humans [5]. Indeed, since 2003 only about 3 in 10 drugs have made it through Phase II testing [8]. If a drug is deemed effective and suitably safe after Phase II, it then moves on to Phase III. Phase III is the largest (and most expensive) of all the phases, with 1000–5000 patients from all over the United States often enrolled in randomized, placebo-controlled experiments [5]. This phase can take up to 10 years to complete, and roughly 10 % of the drugs that make it through Phase II will still fail in Phase III [4]. While less common than failure in Phases I and II, failure in Phase III can be caused due to safety or efficacy shortcomings that only manifest themselves in a large-scale pool of patients, or could simply be a result of the sponsoring institution running out of capital to fund the trial [9]. Success in Phase III depends largely on a drug’s efficacy or safety relative to a “gold standard” (i.e., it has to have some advantage over a market treatment currently used) [5].
If a treatment makes it through Phase III testing, the sponsor can file for a New Drug Application. The FDA will take 6–12 months to review this application; at the end of the review process, they can approve the drug if the Phase III data suggests increased efficacy relative to the gold standard, reject the drug if they believe it to have insufficient safety or to not represent a significant improvement over current treatments, or ask for more data on the drug [4]. An approved NDA means that the new drug can be marketed in the US, after which many drugs will undergo Phase IV trials. Phase IV studies take numerous forms, such as gathering data from consumers and double-blind tests in hospitals that are more similar to Phases II and III testing; these trials are largely concerned with drug safety, especially in populations such as children, which are rarely a part of premarket studies [10]. On the whole, it is estimated that the approval rate for all new oncology drugs entering clinical trials is somewhere around 5 % [7], with oncology drugs having a historically lower than average rate of making it through Phase III [8]. Moreover, an average drug requires somewhere around $1 billion total cost to make it through clinical trials [11].
3 Different Treatments Used with Nanotechnology Cancer Therapeutics
The nanoparticle cancer therapies currently undergoing clinical trials primarily fall into the following three classes of therapeutic mechanisms: chemotherapeutics, short interfering RNA, and imaging agents.
3.1 Chemotherapeutics
While “chemotherapy” is sometimes used as a blanket term to describe any pharmaceutical intervention for cancer, chemotherapeutics are most often considered to be small-molecule drugs that act as purposefully cytotoxic agents. Chemotherapeutics are chosen or designed such that they primarily affect dividing cells, making them aggressive in suppression of the unrestrained cell growth characteristic of cancer [12]. The first chemotherapeutics discovered and employed in human studies were alkylating agents derived from mustard gas used in World War I. Alkylating agents are able to form covalent bonds with DNA, thereby disrupting the mitotic cycle [13]. Since then, many other chemotherapies with alternate mechanisms of action have been developed. Anti-microtubule agents that disrupt the necessary organization of microtubules during cell mitosis, ultimately leading to a disruption in cell division, have been effective at retarding cancer growth. For instance, paclitaxel, which belongs to a class of anti-microtubule agents known as taxanes, inhibits cell growth by stabilizing microtubules, which prevents them from performing the reorganizations necessary for mitosis [14]. Other examples of anti-microtubule agents include dolostatins and rhizoxin [15]. Additionally, cytotoxic antibiotics have been developed as chemotherapeutic agents; while such drugs certainly possess antibacterial properties, they prevent cancer growth by inhibiting cell growth through a variety of mechanisms. Doxorubicin, one of the most commonly used antibacterial chemotherapeutics, binds to several DNA-associated enzymes, thereby damaging DNA and disrupting its replication [16].
Although chemotherapeutics act by impeding cellular division, this effect is not specific to cancer cells. For this reason, chemotherapeutics are associated with many well-known adverse side effects resulting from systemic toxicity. Cancer nanotechnology, therefore, seeks to take advantage of the potent cytotoxicity of chemotherapeutics while limiting the off-target effects that compromise patient well-being. Strategies for doing so include encapsulation of chemotherapeutics in order to improve biodistribution, drug stability, and retention within tumors (e.g., through the EPR effect); complexation with nanoparticles to improve solubility; formulation into nanoparticles with targeting ligands that specifically bind to tumors; and others which are discussed throughout this chapter.
3.2 Short Interfering RNA
Following fundamental studies of RNA interference (RNAi) in C. elegans [17], as well as the studies of the underlying mechanism of RNAi [18], short interfering RNA platforms have been studied intensively as therapeutic agents. Short interfering RNAs, also known as siRNAs, are 21–23 nucleotide double-stranded RNAs that can be synthetically designed to target specific cellular mRNA (Fig. 2). Once in the cytoplasm, these siRNAs are incorporated into an enzyme complex called RISC (RNAi silencing complex), which targets and degrades mRNA complementary to the antisense strand of the siRNA [19]. Properly designed siRNA therapies result in decreased protein expression caused by the mRNA degradation and lead to knockdown of a specific phenotype.
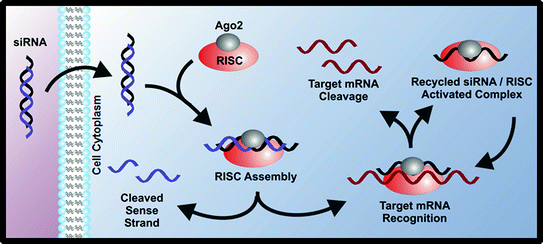
Fig. 2
siRNAs can be used to silence expression of cancer-related proteins through the RNA interference mechanism
Because mRNA degradation is sequence-specific, siRNA has the potential to silence a wide range of target proteins, many of which are “undrugable” and cannot be targeted by small molecule chemotherapeutic agents. Moreover, siRNA is modular: the same base siRNA delivery technology can be applied to different protein targets by changing only the sequence of the siRNA strand. This is especially important for a complex disease like cancer, which can dynamically upregulate multiple oncogenic proteins at once (providing multiple target proteins) and has been known to acquire resistance to some conventional chemotherapeutics [20].
The main hurdle in translation of siRNA lies in the mechanism of delivery; naked siRNA delivered systemically is quickly degraded by various nucleases and/or quickly cleared through the kidneys, making its biodistribution unfavorable and its bioavailability low. Additionally, siRNA is too large and negatively charged to cross cell membranes on its own [21]. To this end, numerous nanoparticle delivery systems that aid in siRNA delivery for treatment of a variety of diseases have been and continue to be studied and developed [22]. Similar to nanoparticles for chemotherapeutics, such particles are intended to enhance siRNA stability, aid in cellular entry, and improve biodistribution. Indeed, siRNA nanoparticle delivery for cancer treatment has been heavily researched, with a few promising technologies having reached clinical trials [23].
3.3 Imaging Agents
In addition to cancer treatments, nanoparticles are also advantageous in diagnosis and imaging of the disease. Given their unique properties, magnetic nanoparticles have the potential to play an important role in the realm of cancer imaging. Superparamagnetic nanoparticles are smaller than the size of a single magnetic domain and, without an applied field, retain no residual magnetization [24]. Thus, these superparamagnetic particles can have their magnetization switched entirely “on” or “off” at will (Fig. 3). When used with magnetic resonance imaging, these nanoparticles confer greater contrast, signal strength, and less aggregation than other imaging agents [25]. As such, several clinical trials utilize nanoparticles as MRI contrast agents. Nanoparticles designed to accumulate in tumors can also serve as optical probes, with fluorescence in the near infrared range used as an alternative to MRI [26].
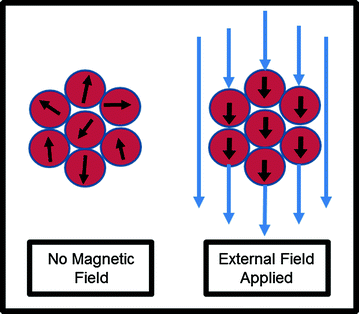
Fig. 3
Superparamagnetic nanoparticles can be used as imaging agents. These nanoparticles can have their magnetization switched “on” with an external field, but retain no residual magnetization without an external field
4 Current Clinical Trials
There are currently many FDA clinical trials involving nanoparticle therapies for cancer, along with a few examples of nanoparticles that have gained FDA approval. The different nanotechnologies that have successfully translated from the lab to clinical trials will be discussed in the following categories: liposomes, polymeric nanoparticles, protein-bound chemotherapeutics, polymer-bound chemotherapeutics, and inorganic nanoparticles.
4.1 Liposomes
Liposomes are spherical nanoparticles made from amphipathic lipids, a class of molecules which have a polar head group and a nonpolar tail group. In aqueous conditions, these lipids naturally self-assemble into a lipid bilayer which forms the outer layer of the liposome (Fig. 4). Drugs can be either entrapped inside the aqueous core or in the hydrophobic membrane, depending on whether the drug is hydrophilic or hydrophobic. Because the encapsulation of the drug in the liposome can reduce drug degradation, limit potential off-target toxicity of the drug, and increase the concentration of the drug at tumors due to the EPR effect, conventionally toxic chemotherapeutics are a natural choice for liposomal formulations. In fact, one of the earliest studies to show the utility of liposomal drugs in vivo used a chemotherapeutic: in 1977, Kobayashi et al. demonstrated significantly enhanced survival of leukemic mice with liposomal ara-C (DNA synthesis inhibitor) compared to ara-C alone [27]. In the decades that followed, liposome technology progressed further with advances such as targeting ligands and triggered release, and several liposomal formulations have made it to the market with hundreds of millions of dollars of sales per year [28].
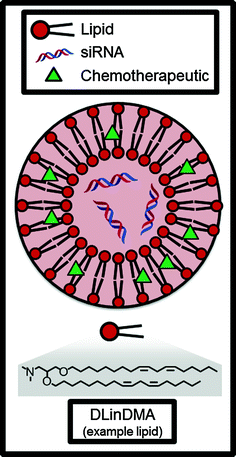
Fig. 4
Liposomes can encapsulate and deliver hydrophobic small molecule chemotherapeutics and/or siRNAs. DLinDMA is an example of an amphiphilic lipid molecule which comprises the bilayer membrane of the nanoparticle
Arguably the most influential liposome technology developed early on was Doxil, a liposomal formulation of the chemotherapeutic doxorubicin. The importance of Doxil to the field of cancer nanotechnology cannot be overstated; as the first FDA-approved nanoparticle, it paved the way for all future generations of liposomal and polymeric particles. For an excellent, firsthand account of the development of Doxil from concept to the clinic, we refer the reader to the review of Barenholz [29]. Briefly, the preclinical development of Doxil was catalyzed by several discoveries, including that (1) PEGylation of the nanoparticle surface increased circulation time and reduced non-specific uptake by macrophages [30] and (2) the use of an ammonium sulfate gradient during drug loading leads to crystallization of the drug and a marked increase in encapsulation efficiency [31]. Pharmacokinetic studies in rats and dogs were conducted [32], and a pilot clinical trial in Israel demonstrated that Doxil localized to the tumor and was overall well-tolerated in humans [33]. Doxil received FDA approval in 1995 for AIDS-related Kaposi’s sarcoma, in 1999 for ovarian cancer, and in 2003 for breast cancer [28]. Doxil is also prolific in clinical trials for other indications or in combination with other therapies, which are too numerous to go into detail here: a search of ClinicalTrials.gov for “Doxil” finds over 1500 registered ongoing or completed clinical trials. Suffice it to say that Doxil’s impact on the field of cancer technology over 20 years after its FDA approval is still enduring today.
After Doxil, many other chemotherapeutics have been incorporated into liposomes and have undergone clinical trials, such as liposomal paclitaxel [34] and liposomal cisplatin [35]; several have also received FDA approval, including DaunoXome (liposomal daunorubicin) and DepoCyt (liposomal cytarabine). Additionally, a non-PEGylated version of liposomal doxorubicin called Myocet has been developed to combat one of the side effects of Doxil, a condition called palmar-plantar erythrodysaesthesia or “hand-foot syndrome” which is characterized by a dermatologic toxic reaction in the hands and feet [36]. There are currently various Myocet formulations in all stages of clinical trials; the most clinically advanced is a Phase III combination therapy for HER2-positive metastatic breast cancer with Mycoet, paclitaxel, and Trastuzumab (Herceptin), an antibody which has been independently shown to improve survival in women with HER2-positive breast cancer [37]. The results of this Phase III trial were published in 2014, and unfortunately, the addition of Myocet was found to not significantly improve the progression-free survival over the standard first-line therapy of paclitaxel and Trastuzumab alone [38]. However, the authors note that Myocet may have benefit and warrants further study in patients whose tumors display a particular subset of biomarkers [38]; because of the great heterogeneity of cancer, it is not uncommon for clinical trials to discover that certain classes of patients respond better to treatment than others.
As an example of a promising new liposome technology, liposomes have recently been developed which are actively targeted to the tumor instead of merely passively accumulating at the site due to the EPR effect. The surface of these liposomes contains antibodies against epidermal growth factor receptor (EGFR), a protein that is overexpressed on the surface of many types of cancer cells [39]. These so-called immunoliposomes delivered cytotoxic drugs more efficiently to EGFR-expressing cells in vitro than liposomes without antibody [40], and this effect was confirmed in vivo with mice [41]. The antibody used on these immunoliposomes was also capable of binding to cells expressing EGFRvIII, a deletion mutant of EGFR often associated with breast carcinoma, non-small cell lung carcinoma, and high-grade glioma. In 2012, the anti-EGFR immunoliposomes became the first targeted nanoparticle delivering chemotherapeutics to undergo a Phase I clinical trial [42]. The doxorubicin-loaded immunoliposomes were administered to patients with EGFR-overexpressing tumors (including pancreatic, head/neck, colorectal, urothelial, and several other cancer indications) in escalating doses, and the recommended dose was determined to be 50 mg/m2 for future Phase II trials; notably, no patients developed palmar-plantar erythrodysaesthesia or cardiotoxicity, which can be associated with Doxil [36, 43].
Liposomes can also be used as a drug delivery vehicle for siRNA therapeutics. Although the most clinically advanced siRNA liposome formulations treat heredity liver disorders (such as Alnylam Pharmaceutical’s patisiran, currently enrolling in a Phase III clinical trial [44]), several siRNA liposome formulations for cancer therapy have recently began or completed Phase I clinical trials. The siRNA entrapped in the aqueous core of these liposomes are designed to silence the expression of genes associated with cancer, including oncogenes (e.g. EphA2) [45] and angiogenesis-promoting genes (e.g., PKN3) [46]. In these lipid nanoparticle formulations, cationic or ionizable lipids are often used because their positive charge (1) improves the entrapment efficiency of the negatively charged siRNA, (2) increases cellular uptake, and (3) facilitates escape of the siRNA from the endosome [22]. The chemical structure of these lipids, which are often composed of an amine-containing polar headgroup and nonpolar hydrocarbon tails, is strongly associated with the potency of the resulting siRNA liposome formulation. Accordingly, thousands of lipid and lipid-like molecules have been synthesized over the past decade using both rational design approaches [47–49] and the synthesis of large combinatorial libraries [50–52] with only a select few of these lipids having sufficient potency and non-toxicity to reach clinical trials. For this reason, liposomes used to encapsulate siRNA are often called “lipid nanoparticles” (LNPs) to highlight the importance of the lipid itself.
Reported in the mid-2000s, one of the first lipids to be developed for use in siRNA-LNP formulations was DLinDMA (Fig. 3) [47]. DLinDMA and siRNA can be formulated into nanoparticles along with phospholipid (to increase efficacy), cholesterol (to increase stability), and lipid-anchored PEG (to increase LNP circulation time, reduce aggregation, and reduce nonspecific uptake by macrophages) [22]. Because it contains these additional excipients, the nanoparticle is classified as a SNALP (stable nucleic acid lipid nanoparticle); SNALPs have been shown to typically distribute to the liver in vivo and increase efficacy to hepatocytes over simple lipid-siRNA lipoplexes [53]. In 2006, DLinDMA SNALPs were used to demonstrate the first RNAi-mediated gene silencing in non-human primates [54], and this technology was then translated to the clinic by Alnlyam Pharmaceuticals, which in 2011 completed a Phase I dose-escalation clinical trial [55]. The DLinDMA SNALP was called ALN-VSP02 and incorporated siRNAs targeting both vascular endothelial growth factor (VEGF, promotes angiogensis) and kinesin spindle protein (KSP, promotes cellular proliferation) for the treatment of solid tumors. Published results from this Phase I clinical trial indicate that intravenously-administered ALN-VSP02 was generally well-tolerated at doses ranging from 0.1 to 1.5 mg/kg with no clinically significant liver toxicity in 40 of 41 patients; additionally, one patient with nodal and extensive liver metastases from endometrial cancer achieved a complete response to the therapy and three patients (with both hepatic and extrahepatic metastases) achieved stable disease for 1–1.5 years [56].
Over the past decade, the chemical structures of the lipids have been optimized by different researchers to reduce toxicity, change the biodistribution, and reduce the IC50 of the siRNA-LNPs. (A lower IC50 is desirable because it reduces the amount of lipid required and thus may alleviate concerns of any potential long-term toxicity of the lipid.) A lipid called DLin-MC3-DMA from Tekmira was incorporated into a formulation called TKM-PLK1 designed to treat primary or secondary liver cancer, and it has recently completed Phase I clinical trials [48, 57]. The siRNA encapsulated in TKM-PLK1 targets polo-like kinase 1 (PLK1), a protein frequently overexpressed in tumors and associated with carcinogenesis [58]. A cationic lipid called AtuFECT01 from Silence Therapeutics which targets the endothelium [59] was incorporated into a formulation called Atu027. Atu027 contained siRNA targeting protein kinase 3 (PKN3, promotes angiogenesis) which retarded tumor growth in preclinical mouse studies [46], was well-tolerated in Phase I trials for patients with advanced solid tumors with some anti-metastatic activity observed [60, 61], and is currently enrolling in a Phase1b/IIa study in patients with pancreatic adenocarcinoma [62].
TKM-PLK1, ALN-VSP02, and Atu027 have completed Phase I clinical trials, but there are additional siRNA-LNP formulations for cancer therapy on the horizon. A Phase I clinical trial has been registered which uses the neutrally-charged lipid 1,2-dioleoyl-sn-3-phosphatidylcholine (DOPC) to encapsulate siRNA targeting epithelial cell kinase (EphA2), an oncoprotein overexpressed in many types of human tumors and especially ovarian tumors [45, 63]. Moreover, Dicerna Pharmaceuticals is currently enrolling patients for a Phase I clinical trial for the treatment of solid tumors, multiple myeloma, and non-Hodgkins lymphoma with their LNP formulation called DCR-MYC [64]. DCR-MYC is made from a proprietary lipid called EnCore and contains 27-nucleotide siRNA targeting the oncogene myc, which is frequently overexpressed in cancers [65–67].
4.2 Polymeric Nanoparticles
In contrast with LNPs, polymers can also be used to form spherical polymeric micelles to encapsulate and deliver chemotherapeutics. These nanoparticles are distinguished from polymer-bound chemotherapeutic conjugates (discussed in a later section) because the drug here is encapsulated in the particle rather than covalently bonded to the polymer, although it should be noted that some conjugates will spontaneously form nanoparticle-like shapes in aqueous conditions [68]. Polymer micelles are made when an amphiphilic block copolymer self-assembles in an aqueous solution, trapping the hydrophobic drug in the hydrophobic inner micelle core (Fig. 5).
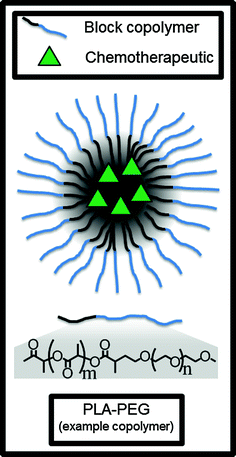
Fig. 5
Polymer micelles can encapsulate and deliver hydrophobic small molecule chemotherapeutics in the hydrophobic core. PLA-PEG is an example of an amphiphilic block copolymer which self-assembles into the micelle in aqueous conditions
The formulation called Genexol PM (also called cynviloq or IG-001) from Sorrento Therapeutics uses a micelle made from a PEG (hydrophilic) and poly(D,L-lactic acid) (hydrophobic) block copolymer to encapsulate paclitaxel and release the drug in a controlled fashion [69]. Genexol PM is approved for clinical use in South Korea to treat metastatic breast cancer and non-small cell lung cancer (NSCLC), is undergoing Phase II clinical trials in the United States for various other indications like urothelial cancer [70], and is currently underway in a bioequivalency crossover clinical trial with albumin-bound paclitaxel as the control alternative therapy [71]. According to a Sorrento press release [72], Genexol PM may be approved by the FDA under the 505(b) [46] pathway for a new drug application if it demonstrates equivalent pharmacokinetics to albumin-bound paxlitaxel. Several other polymeric micelle-based nanoparticles are in Phase III clinical trials. For example, NK105, made from a PEG/modified polyaspartate block copolymer and encapsulating paclitaxel [73], is now in a Phase III clinical trial for the treatment of breast cancer [74]. NK105 was also well-tolerated with 14/56 patients having a clinical response in a Phase II trial for gastric cancer [75].
Targeting moieties are also being introduced to the polymeric nanoparticle surface to deliver the particle to tumor cells with better specificity. One example of this concept is Bind Therapeutic’s BIND-014, a nanoparticle made from a PEG/poly(lactic-co-glycoclic acid) block copolymer encapsulating docetaxel. The surface of BIND-014 is decorated with a small molecule called ACUPA which has been shown to have specificity for prostate specific membrane antigen (PSMA), a receptor overexpressed on prostate cancer cells and solid tumor vasculature [76]. In preclinical development, BIND-014 was selected and optimized from a large combinatorial library of nanoparticles with varying sizes, polymer composition, ACUPA targeting molecule density, and other parameters [77]. It was reported that 2/12 patients had tumor shrinkage in a Phase I dose escalation clinical trial [77, 78], and BIND-014 is currently being studied in two Phase II clinical trials for prostate cancer [79] and NSCLC [80].
Polymer nanoparticles have predominately been used to deliver small molecule chemotherapeutics but can also be used to deliver siRNA for cancer treatment. First described in 1999 by Davis and coworkers, β-cyclodextrin is a sugar-derived, linear, cationic polymer which can form nanoparticle complexes with negatively charged siRNA [81]. The nanoparticle formulation called CALAA-01 is made of β-cyclodextrin, PEG, and siRNA targeting ribonucleotide reductase subunit M2 (RRM2, an enzyme involved in DNA replication); furthermore, the surface of CALAA-01 nanoparticles is decorated with the targeting moiety transferrin, a protein which binds to transferrin receptors overexpressed on cancer cells [82]. In 2008, a Phase I clinical trial for CALAA-01 began, the first clinical trial for targeted siRNA nanoparticles [83]. An interim publication released in 2010 investigated the tumor biopsies of some of the patients and discovered the first evidence of siRNA-mediated mRNA degradation in humans [84], an important proof-of-concept for the mechanism of RNAi and a milestone in the gene delivery field. The Phase I trial was terminated in 2013, and to our knowledge no explanation for this termination has been given by Arrowhead Research Corporation. However, the β-cyclodextrin polymer technology has seen better success in polymer-bound chemotherapeutics as discussed in a subsequent section.
4.3 Albumin-Bound Chemotherapeutics
By numbers alone, the most common types of cancer-treating nanoparticle currently undergoing clinical trials are albumin-bound chemotherapeutics. Specifically, these platforms utilize albumin to form a nanoparticle carrier for chemotherapeutic drugs. Because albumin is a natural carrier of hydrophobic materials, it can be used to overcome solvation issues with highly hydrophobic drugs that would otherwise need to be solubilized and delivered using harmful solvents. Additionally, albumin is known to interact with the cellular GP60 receptor, which improves the trafficking of the bound drug into the cell [85]. These nanoparticle albumin bound (nab) platforms therefore overcome the dose restrictions on potent chemotherapeutic agents (such as taxanes) dissolved in cytotoxic solvents.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


