in hematopoietic cells at all stages of differentiation. It can localize to the mitochondria during oxidative stress. It is the human homolog of the Abelson B-cell murine leukemia virus oncogene (v-abl). The ABL1 protein participates in signal transduction and regulation of gene transcription. One major function is to catalyze the attachment of phosphate groups to the tyrosine residues of various proteins, that is, to act as a TK. Members of the TK family are frequently involved in the pathways that transmit signals from the external milieu to the cytoplasm and nucleus; in this capacity, they may act as growth factors, transmembrane receptors, or submembrane catalytic subunits of surface receptors.
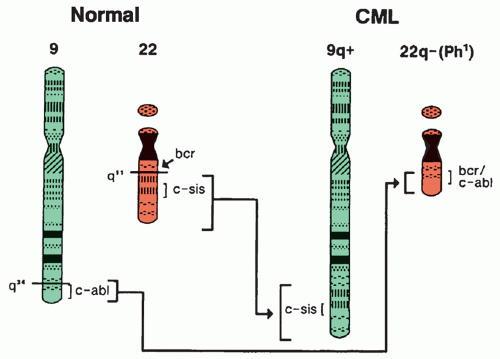 Figure 21.1 Anatomy of the (9;22)(q34;q11) translocation with formation of the Philadelphia (Ph1) chromosome (22q-) containing the hybrid BCR-ABL1 gene. (See text for further explanation.) CML, chronic myelocytic leukemia. |
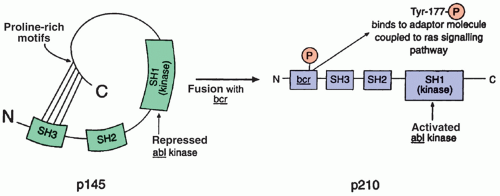 Figure 21.2 The normal abl tyrosine kinase (TK) component of P145 is carefully regulated by the SH2 and SH3 domains; genetic alterations that prevent interaction of the SH3 domain with proline-rich motifs within the C terminus result in derepression (constitutive activation) of the TK. In Philadelphia chromosome-positive chronic myelocytic leukemia, fusion with the bcr gene leads to production of a chimeric protein (P210) with activated TK and oncogenic properties. Two domains within bcr are required for its oncogenic effect: domain I mediates oligomerization of bcr/abl and promotes phosphorylation of tyrosine residue 177 within domain II, which, in turn, binds to a signaling adaptor molecule coupling bcr/abl with the ras signaling pathway. (See text for further explanation.) |
TABLE 21.1 Functional Domains of ABL, BCR, and BCR-ABL1 Proteins | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TK (SH1) domain. By engaging the C-terminal lobe of SH1, it facilitates the docking of the SH2 and SH3 domains onto it, thereby blocking the access of ATP and the peptide substrate to the active site and inactivating the TK activity. In contrast, forms of ABL1 that lack myristoylation (or replace the myristoylated Cap with unmyristoylated BCR) manifest constitutive TK activity. (reviewed in Ref. 9)
 Figure 21.3 Schematic representation of the normal protein product (P145) of the ABL1 gene and the modifications associated with leukemogenesis. Note that all variants retain the tyrosine kinase domain (blue area) and are altered at the 58 end by insertion of the viral gag component in the case of Abelson murine leukemia virus (AbMuLV) and feline sarcoma virus (FeSV) or with BCR segments in the case of Philadelphia chromosome-positive (Ph1-positive) ALL and chronic myelocytic leukemia (CML). |
patients were previously all classified as Philadelphia chromosome-negative CML; however now this heterogeneous population of patients can be better categorized as distinct Ph1-negative myeloproliferative neoplasms based on clinical characteristics and genetic features.1
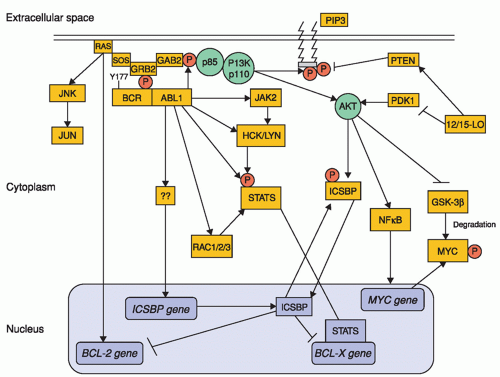 Figure 21.4 Molecular signaling in BCR-ABL1-positive myeloid precursors. The phosphorylated Tyr177 residue of BCR serves as a docking site for growth factor receptor-bound protein 2 (GRB2), which binds GRB2-associated binding protein 2 (GAB2), as well as SOS (a guanine nucleotide exchanger of RAS), resulting in RAS-MAPK activation; this, in turn, promotes transcription of BCL-2 gene. Upon phosphorylation, GAB2 recruits phosphatidylinositol 3-kinase (PI3K), which activates AKT. AKT activation increases transcription of MYC gene and stabilizes MYC protein via inhibition of its degradation by GSK-3β. BCR-ABL-1 also activates STAT5, both directly and indirectly through activation of JAK2 and the SRC kinases HCK and LYN. The end result is the activation of BCL-X gene transcription. In addition, BCR-ABL1 abrogates the transcription of interferon consensus sequence-binding protein (ICSBP), thereby releasing the ISCBP-mediated inhibition of BCL-2 and BCL-X gene transcription, resulting in increased survival of myeloid precursors. Thus, the net effect of BCR-ABL1 kinase activation is the promotion of cell proliferation and survival. Pointed arrows indicate direct interactions and/or activation. Blunt-ended arrows indicate inhibitory effects. GSK-3β, glycogen synthase kinase-3β; PIP3, phosphatidylinositol-3,4,5 triphosphate. (From Quintos-Cardama A, Cortes J. Molecular biology of bcr-abl-positive chronic myeloid leukemia. Blood 2009;113:1619, used with permission.) |
TABLE 21.2 Relationship of BCR Domains to BCR/ABL1 Fusion Genes | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
basophils, minimal absolute monocytosis (<10%), hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia and <20% blasts in the blood and marrow. Chromosomal changes have been reported in 20% to 88% of aCML cases, most commonly, trisomy 8 and del (20q). Recently, much has been learned about the molecular basis of this rare disease through next generation sequencing efforts. Non-mutually exclusive mutations in the receptor for colony-stimulating factor 3 (CSF3R), the Set binding protein, SETBP1, and JAK2V617F have been identified in 40%, 25%, and 10% of aCML patients, respectively. The identification of such mutations may be useful as a diagnostic criterion for aCML,23,24 and may have therapeutic implications. CSF3R mutations signal through JAK or SRC kinase pathways and thus JAK inhibitors such as ruxolitinib or SRC kinase inhibitors such as dasatinib may prove therapeutically useful.25
CCR7.43 This defective adhesion to the bone marrow stroma may lead to release of immature cells into the circulation and may facilitate hematopoiesis in extramedullary sites.
TABLE 21.3 Criteria for Chronic, Accelerated, and Blast Phases of CML | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
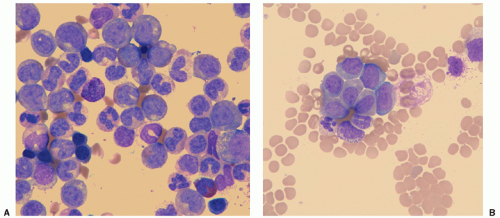 Figure 21.5 Peripheral blood smears of chronic myelocytic leukemia. A: Chronic phase—marked leukocytosis showing the entire range of myeloid cells from myeloblast to mature polymorphonuclear leukocytes; a hypergranular eosinophil and basophil are present as well. B: Blast phase—blast cells are now more prominent, and there is a hiatus in myeloid maturation. (Courtesy of Drs. Hiroko Shinoda and William Rezuke.) |
and eosinophils is noted. Hybrid eosinophilic-basophilic granulocytes may also be seen46; because similar chimeric granules may be found in normal immature granulocytes, this phenomenon may reflect incomplete maturation. The mean platelet count in children is approximately 500,000/mm3, which is not significantly higher than that in adults.6,7 Serologic findings include elevation of uric acid, lactate dehydrogenase, vitamin B12, and vitamin B12-binding protein (transcobalamin 1).
TABLE 21.4 Criteria for Response to CML Therapy | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
specific suppressor of the neoplastic clone.63,64 Imatinib was approved by the United States Food and Drug Administration (FDA) for adults with CP CML in 2001 and for children with CP CML in 2003. Since then, three second-generation TK inhibitors (TKIs), dasatinib, nilotinib, and bosutinib have also received FDA approval for adult patients resistant to or intolerant of imatinib. With additional adult studies demonstrating the superiority of dasatinib (DASISION study) and nilotinib (ENESTnd study) over imatinib, therapeutic recommendations issued by both the National Comprehensive Cancer Network and the EuropeanLeukemiaNet include the upfront use of imatinib, dasatinib, or nilotinib for adult patients in CP.56,65,66,67
TABLE 21.5 Results of Treatment of Chronic Phase CML | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
assessed in newly diagnosed children (ages 2 to 19 years) in CP.73 CHR was attained by 80% of patients by 2 months of therapy. CCyR was achieved in 38% of patients by 3 months and overall in 72% of patients at a median time of 5.6 months. Ninety-one percent of the patients who achieved CCyR did so by 9 months of therapy. The rate of CMR was 27%. Progression-free and overall survival at 3 years were 72% and 92%, respectively.73 A French multicenter phase IV trial treated 44 newly diagnosed children with CP CML with 260 mg/m2 imatinib. At 36 months, PFS was 98%. The rates of CCyR at MMR at 12 months were 61% and 31%, respectively. Cumulative CCyR and MMR rates were 77% and 57%, respectively.74
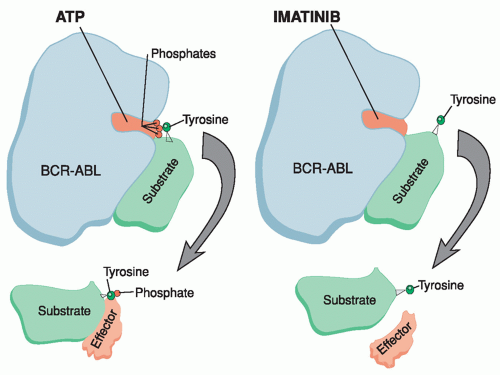 Figure 21.6 Mechanism of action of imatinib mesylate. In the untreated state (left), BCR-ABL protein has an open pocket accessible to ATP. This facilitates transfer of a phosphate moiety from ATP to a tyrosine residue on a target substrate molecule. The activated molecule is then released to interact with downstream effector molecules, which can promote oncogenesis. Imatinib inhibits this process by competing with ATP for the kinase pocket, thereby preventing phosphorylation of substrate and effector molecules. (Courtesy of Eleanore Rhodes.) |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


