Myeloproliferative Neoplasms and Thrombohemorrhagic Complications
Myeloproliferative Neoplasms and Thrombohemorrhagic Complications
Ayalew Tefferi
The term “myeloproliferative disorder” (MPD) was first introduced by Dameshek1 in 1951 to emphasize the clinicopathologic interrelationship between essential thrombocythemia (ET), polycythemia vera (PV), primary myelofibrosis (PMF), chronic myeloid leukemia (CML), and erythroleukemia (di Guglielmo’s syndrome). Between 1967 and 1981, Fialkow et al. showed that the “MPDs” were also biologically interrelated on the basis of being clonal stem cell disorders with involvement of both myeloid and lymphoid lineage.2, 3, 4, 5, 6, 7, 8, 9 In the interim, several other myeloid malignancies have been described and shown to display clinicopathologic as well as biologic attributes that are similar to those of Dameshek’s “MPD.” Furthermore, there is now plenty of evidence to consider MPDs as myeloid malignancies and the name has accordingly been changed to “myeloproliferative neoplasms (MPNs).”
The World Health Organization (WHO) system recognizes five categories of myeloid malignancies including acute myeloid leukemia (AML), myelodysplastic syndromes (MDSs), MPNs, MDS/MPN overlap, and PDGFR/FGFR1-rearranged myeloid/lymphoid neoplasm with eosinophilia (Table 102.1).10 “BCR-ABL1-negative MPN” is an operational subcategory of MPN that includes PV, ET, and PMF.11 All three disorders are characterized by stem cell-derived clonal myeloproliferation and the presence of somatic mutations involving primarily JAK2 and to a lesser extent MPL, LNK, CBL, TET2, ASXL1, IDH, IKZF1, EZH2, DNMT3A, TP53, or SF3B1 mutations.12, 13, 14, 15, 16, 17 None of these mutations appear to garner the disease specificity or pathogenetic relevance otherwise displayed by BCR-ABL1, in CML.12
EPIDEMIOLOGY
Incidence figures for ET, PV, and PMF vary across studies but a population-based report from Olmsted county, Minnesota, USA provided estimates of 2.5, 1.9, and 1.5/100,000, respectively.18, 19 The corresponding figures for median age at diagnosis were 72, 67, and 70 years. All three BCR-ABL1-negative MPNs are rare in children and there is a slight male predominance in PMF and female predominance in ET.20, 21, 22 A higher disease incidence involving all three MPNs has been suggested in persons of Jewish ancestry.23, 24, 25, 26, 27 In general, there is no hard evidence that links any of these MPNs with environmental toxins.
Clinical Features
Most patients with ET are currently diagnosed incidentally and in an asymptomatic state.28 However, symptoms and signs of disease are usually present when either PV or PMF is diagnosed.29, 30 Thrombohemorrhagic manifestations are the most frequent life threatening events in both PV and ET, and include microvascular disturbances (headache, lightheadedness, visual symptoms, palpitations, atypical chest pain, acral paresthesias, erythromelalgia), thrombotic episodes (stroke, transient ischemic attacks, retinal vein occlusion, cerebrovascular vein thrombosis, myocardial infarctions, angina pectoris, pulmonary embolism, abdominal large vein thrombosis, extremity deep vein thrombosis, digital ischemia, gangrene), and bleeding complications (central nervous system, retinal, mucosal, gingival, gastrointestinal, retroperitoneal, cutaneous, deep tissue, post-surgical, aspirin induced).28, 31, 32, 33 Bleeding in both PV and ET is exacerbated by the use of aspirin and other nonsteroidal antiinflammatory drugs.
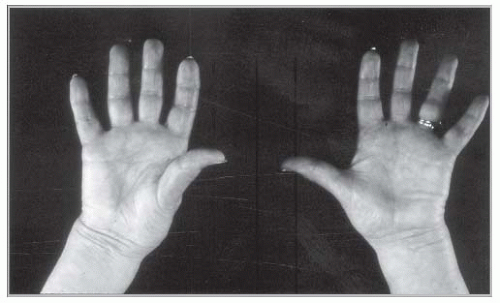
Microvascular symptoms, especially erythromelalgia, are the result of small vessel platelet-endothelium interaction with associated inflammation and transient thrombotic occlusion.34 Erythromelalgia represents acral erythema, warmth, and pain (FIGURE 102.1).35 Young women with ET may present with recurrent first trimester miscarriages.36 Newly diagnosed or untreated patients with PV might display plethora (a red and congested facial complexion), palmar erythema, and sausage-shaped distention of retinal veins. They might also experience symptoms of hyperviscosity that include head fullness, dizziness, flushing, visual disturbances, tinnitus, epistaxis, dyspnea, and increased blood pressure.37, 38, 39 Nonvascular symptoms in PV include pruritus and constitutional symptoms including fatigue, malaise, and night sweats. PV-associated pruritus occurs in more than 50% of patients and is usually exacerbated by taking a bath.40 Other symptoms and signs of PV include gout, splenomegaly, and early satiety.
The typical clinical presentation in PMF includes progressive anemia, severe constitutional symptoms, and marked splenomegaly.30 Hepatosplenomegaly in PMF is secondary to extramedullary hematopoiesis (EMH). EMH in PMF may also occur in nonhepatosplenic tissue including lymph nodes, pleura (effusion), peritoneum (ascites), lung (interstitial process), and the paraspinal and epidural spaces (spinal cord and nerve root compression). Constitutional symptoms in PMF include profound fatigue, weight loss, night sweats, and low-grade fever. Weight loss in PMF consists of mostly muscle mass wasting. Other disease manifestations include peripheral edema, diarrhea, early satiety, portal hypertension, and splenic infarcts.41, 42
Table 102.1 WHO classification of myeloid malignancies
1. Acute myeloid leukemia (AML) and related precursor neoplasmsa
3.1. Refractory cytopeniab with unilineage dysplasia (RCUD)
3.1.1. Refractory anemia (ring sideroblasts <15% of erythroid precursors)
3.1.2. Refractory neutropenia
3.1.3. Refractory thrombocytopenia
3.2. Refractory anemia with ring sideroblasts (RARS; dysplasia limited to erythroid lineage and ring sideroblasts ≥15% of bone marrow erythroid precursors)
3.3. Refractory cytopenia with multilineage dysplasia (RCMD; ring sideroblast count does not matter)
3.4. Refractory anemia with excess blasts (RAEB)
3.4.1. RAEB-1 (2%-4% circulating or 5%-9% marrow blasts)
3.4.2. RAEB-2 (5%-19% circulating or 10%-19% marrow blasts or Auer rods present)
4.4.1. Provisional entity: Refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T)
5. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA,c PDGFRB,c or FGFR1c
5.1. Myeloid and lymphoid neoplasms with PDGFRA rearrangement
5.2. Myeloid neoplasms with PDGFRB rearrangement
5.3. Myeloid and lymphoid neoplasms with FGFR1 abnormalities
a AML-related precursor neoplasms include “therapy-related MDS” and “myeloid sarcoma.”
b Either mono- or bicytopenia: hemoglobin level < 10 g/dL, absolute neutrophil count < 1.8 × 109/L, or platelet count < 100 × 109/L. However, higher blood counts do not exclude the diagnosis in the presence of unequivocal histologic/cytogenetic evidence for MDS.
c Genetic rearrangements involving platelet-derived growth factor receptor α/β (PDGFRA/PDGFRB) or fibroblast growth factor receptor 1 (FGFR1).
FIGURE 102.1 Erythromelalgia represents painful, erythematous discoloration of the hands or toes associated with warmth and burning sensation.
PATHOGENESIS
MPNs Are Clonal Stem Cell Diseases with Recurrent Somatic Mutations
As early as the 1970s and 1980s, the use of glucose-6-phosphate dehydrogenase (G-6-PD) isoenzyme analysis has suggested that patients with MPN including ET,9 PV,4 and PMF7 have clonal hematopoiesis that originates at the stem cell level and may involve both myeloid and B lymphoid lineage.43 This early observation has been supported by more recent investigations using X-linked DNA44, 45 and transcript46 analysis in informative females. The disease-causing events in BCR-ABL1-negative MPN remain elusive. Beginning in 2005, several somatic mutations involving primarily JAK2 and to a lesser extent MPL, LNK, CBL, TET2, ASXL1, IDH, IKZF1, EZH2, DNMT3A, TP53, or SF3B1 have been described.12, 13, 14, 15, 16, 17 However, none of these mutations appear to garner the disease specificity or pathogenetic relevance otherwise displayed by BCR-ABL1, in CML.12 Among the aforementioned mutations, JAK2 and MPL have been more closely associated with MPN.
JAK2V617F results from a somatic G to T mutation involving JAK2 exon 14, which leads to nucleotide change at position 1849 and the substitution of valine by phenylalanine at codon 617.47 The mutation affects the JH2 domain and is believed to derail its kinase-regulatory activity. JAK2V617F-mediated transformation is believed to require co-expression of type 1 cytokine receptor and leads to STAT5/3 activation.48, 49, 50, 51 Also, JAK2V617F might have an epigenetic effect through nuclear translocation of the mutant molecule and direct phosphorylation of histone H3.50JAK2V617F induces PV-like phenotype in mouse transplantation models52 and this observation has been further confirmed by a recent report of an inducible JAK2V617F knock-in mouse model where both heterozygous and homozygous mutation expression induced PV-like disease, with the latter causing a more aggressive phenotype with myelofibrosis (MF).53 Such experimental data along with the fact that virtually all patients with PV carry a JAK2 mutation54 suggest a cause-effect relationship with erythrocytosis.55, 56, 57, 58, 59JAK2V617F homozygosity is infrequent in ET and its frequent occurrence in PV has been ascribed to mitotic recombination, possibly facilitated by JAK2V617F-induced genetic instability.60
JAK2V617F-positive MPN has been associated with older age at diagnosis (ET and PMF), higher hemoglobin level (ET and PMF), leukocytosis (ET and PMF), and lower platelet count (ET).61 A higher mutant allele burden has been associated with pruritus (PV and PMF), higher hemoglobin level (PV), leukocytosis (PV, ET, and PMF), and larger spleen size (PV, ET, and PMF).62, 63, 64, 65, 66 However, the mere presence of JAK2V617F or increased mutant allele burden does not appear to affect survival or leukemic transformation.66, 67, 68, 69, 70, 71, 72JAK2V617F allele burden increases with time in PV and PMF,63, 65, 73 but not in ET.66 Current evidence is not conclusive with regard to the relationship between JAK2V617F and thrombosis.65, 66, 67, 68, 74, 75, 76JAK2 exon 12 mutations are relatively specific to JAK2V617F-negative PV.55JAK2 exon 12 mutation-positive PV patients are often heterozygous for the mutation and are characterized by predominantly erythroid myelopoiesis, subnormal serum erythropoietin (Epo) level, and younger age at diagnosis.55, 77, 78 The clinical course of these patients appears to be similar to that of patients with JAK2V617F-positive PV.56, 78, 79
MPL (Myeloproliferative Leukemia Virus oncogene) is located on chromosome 1p34. Gain-of-function germline MPL mutations have been associated with familial thrombocytosis (S505N) that is, interestingly, associated with an MPN-phenotype including splenomegaly, MF, and an increased risk of thrombosis.80 An MPL single nucleotide polymorphism (G1238T) that results in a K39N substitution is found in approximately 7% of African Americans and is associated with higher platelet counts.81 Somatic MPL mutations are rare and their occurrence is largely limited to patients with MPN. MPLW515L results from a G to T transition at nucleotide 1,544 (exon 10) resulting in a tryptophan to leucine substitution at codon 515. MPLW515L was first described in patients with JAK2V617F-negative PMF and induces a PMF-like disease with thrombocytosis in mice.82 Subsequently, MPLW515K and other exon 10 MPL mutations (e.g., MPLW515S and MPLS505N) were described in ET and PMF with mutational frequencies that range from 3% to 15%.82, 83, 84, 85, 86, 87
MPL-mutated ET has been associated with older age, lower hemoglobin level, higher platelet count, microvascular symptoms, and a higher risk of post-diagnosis arterial thrombosis.87, 88 The presence of MPL mutation did not appear to affect survival, fibrotic, or leukemic transformation.87MPL-mutated PMF has been associated with the female gender, older age, lower hemoglobin level, and a higher likelihood of becoming transfusiondependent.86 MPL mutations do not affect survival or leukemic transformation in PMF.89
MPNs Are Associated with Bone Marrow Stromal Reaction
Bone marrow histology in MPN, primarily in PMF,90, 91 but also to some extent in both PV92, 93 and ET,94, 95, 96 displays excess collagen fibrosis, new bone formation, and angiogenesis.95, 97, 98, 99, 100, 101 Bone marrow fibroblasts in MPN are polyclonal and participate in the altered cellular as well as extracellular concentrations of various fibrogenic and angiogenic cytokines.102, 103, 104, 105 The latter are believed to derive from clonal megakaryocytes and/or monocytes and include platelet-derived growth factor (PDGF), transforming growth factor-β, basic fibroblast growth factor, and vascular endothelial growth factor. The current assumption is that these as well as other cytokines mediate the bone marrow stromal reaction in MPN.102, 103, 104 Furthermore, increased tissue levels of pathogenetic cytokines might be promoted by an altered interaction between megakaryocytes and neutrophils that is promoted by increased expression of P-selectin by the former.106 Similarly, neutrophil-derived elastase and other enzymes might contribute to the abnormal peripheral blood egress of myeloid progenitors in PMF.107, 108
MPNs Are Associated with Endogenous Myeloid Colony Growth
Endogenous (spontaneous, growth factor-independent) in vitro erythroid or megakaryocyte colony formation is not seen in either normal subjects or reactive myeloproliferation.109 Epoindependent erythroid proliferation is primarily seen in PV,110, 111, 112 but is also seen in a proportion of patients with ET113, 114, 115, 116 or PMF.114, 117 Furthermore, endogenous colony growth is also manifested by granulocyte118 and megakaryocyte119 progenitors. Such growth factor-independence has not been attributed to mutations in ligand receptor120, 121 or receptor-associated signal transducer molecules.122
In Vitro Myeloid Growth in MPN is Associated with Growth Factor Hypersensitivity
Growth factor hypersensitivity under in vitro conditions has been demonstrated for both PV and ET. Erythroid cells in PV are hypersensitive to a variety of cytokines including insulin-like growth factor (IGF-1),123 stem cell factor,124 granulocyte-monocyte colony-stimulating factor,125 interleukin-3 (IL-3),126, 127 and Tpo.128 A similar growth factor hypersensitivity to IL-3129 or Tpo130 has also been suggested in ET. The consistently observed IGF-1 hypersensitivity of erythroid cells in PV has been attributed to alterations in IGF-1 binding proteins.131, 132 The receptors for both Epo120, 133, 134 and Tpo121, 135 have been examined in patients with MPN and found to be intact.
MPNs Are Associated with Decreased Megakaryocyte/Platelet Mpl Expression
Decreased megakaryocyte/platelet Mpl expression was first reported in ET136 and subsequently in PV137 and other MPNs.138, 139 In ET, the decreased surface expression of Mpl has been associated with reduced c-Mpl transcription.136, 140 In PV, the particular defect was traced to a post-translational hypoglycosylation of the Mpl protein with derailment of membrane localization.141
Pathogenesis of Bleeding Diathesis in MPN
Platelet morphology in MPN is variable and includes both small and giant forms.142 These platelets are often hypogranular, and on electron microscopy display decreased granular contents involving both δ (serotonin, adenine nucleotides) and a (PDGF, β-thromboglobulin, platelet factor 4) specific storage granules.143, 144, 145, 146, 147, 148 This acquired storage pool deficiency as well as altered surface expression of hemostatic molecules is believed to contribute to the prohemorrhagic platelet defects in MPN.
In vitro observed hemostatic abnormalities in MPN include poor platelet aggregation in response to various platelet agonists (i.e., thrombin, ADP, epinephrine, collagen, thromboxane A2 (TXA2), platelet-activating factor),149, 150, 151, 152 abnormally low intra-platelet levels of adenine nucleotides and serotonin,153 reduced platelet factor X-activating activity,154 defective platelet lipid peroxidation,155 impaired binding to fibrinogen as a result of decreased glycoprotein IIb/IIIa expression,156 decreased adrenergic receptor expression that could explain the often observed absent epinephrine response and mechanistically involve impaired signal transduction,157, 158 and acquired von Willebrand syndrome (AvWS).159 Similarly, quantitative radiolabeled as well as flow cytometric studies in PV and ET have revealed decreased expression of platelet glycoprotein receptors Ib and IIb/IIIa.156, 160, 161
AvWS occurs in more than a third of the patients with MPN and has been associated with a bleeding diathesis.162, 163 Several investigators have established a relationship between the platelet count and loss of large von Willebrand factor (vWF) multimers in plasma of MPN patients.164, 165, 166 The finding that large vWF multimers in plasma decrease progressively at increasing platelet counts of >1,000 × 109/L is in agreement with epidemiologic studies in ET showing an increased bleeding risk when the platelet count exceeds this level.31, 167, 168, 169 The functional deficiency of MPN-associated acquired von Willebrand disease (AvWD) may not be apparent when measuring vWF:Ag and FVIII levels alone, as these are typically maintained within the normal range.159, 170
The assays used to assess vWF function (collagen binding activity [VWF:CBA] or ristocetin cofactor activity [vWF:RCoA]) reveal that vWF function declines relative to the vWF:Ag with increasing platelet counts.171, 172, 173 This phenomenon is not restricted to MPN, but has also been described among patients with reactive thrombocytosis (RT).164 Therefore, the observed loss of large multimers in ET and PV is likely an effect of the absolute platelet number, rather than an effect of dysfunctional clonal platelets. Further evidence of the importance of platelet number is provided by the observed resolution of the clinical bleeding complications and the AvWD in ET and PV patients following platelet cytoreduction with myelosuppressive therapy or platelet pheresis.159, 164, 170, 172 Similarly, resolution of RT also restores the large multimers and, as a result, vWF function among affected patients.164
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



