General Overview
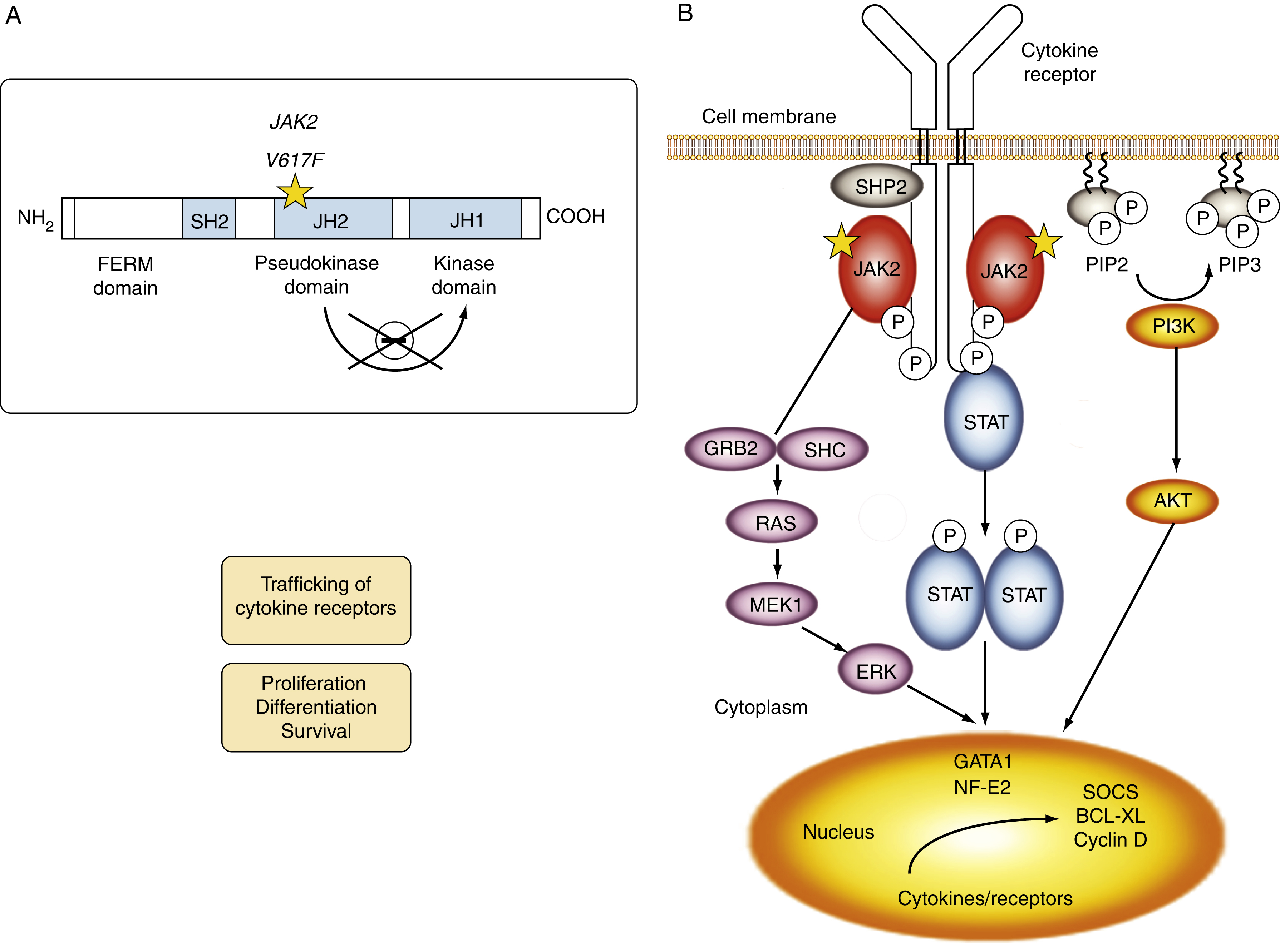
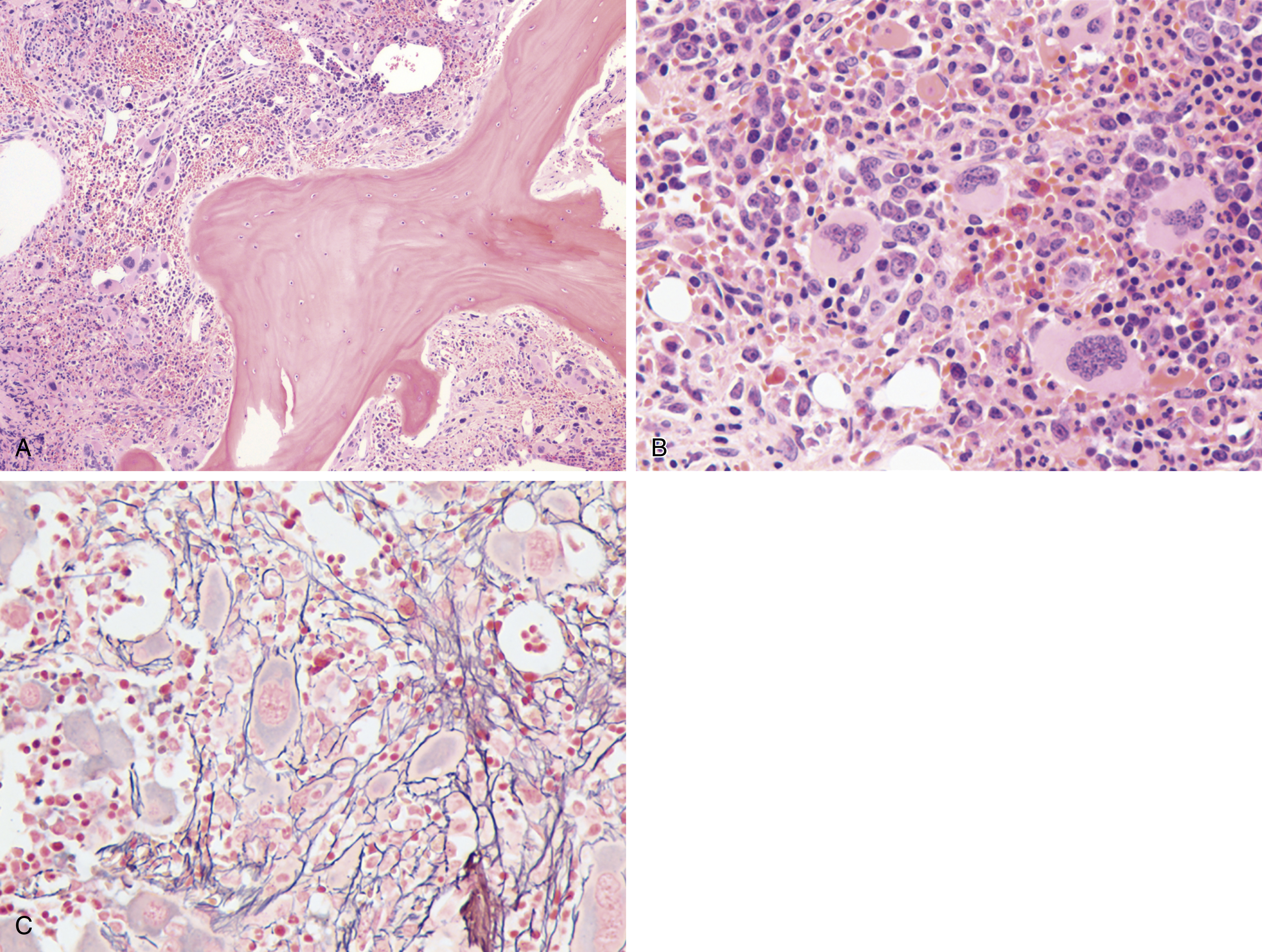
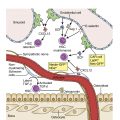
Myeloproliferative neoplasms (MPNs) are clonal stem cell disorders characterized by abnormal chronic proliferation of cells belonging to myeloid lineages that are often associated with elevated blood counts, splenomegaly, and a propensity for thrombosis. The World Health Organization (WHO) 2016 classification recognizes MPNs as myeloid neoplasms, and the three classic Philadelphia chromosome (Ph)–negative MPNs are polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). Both PV and ET carry a risk of transformation to post-PV or post-ET MF, which carry the same risks and symptoms as PMF and are treated in the same manner. Bone marrow (BM) fibrosis grade, marrow, and bony changes are shown in Figs. 5.1 through 5.3 . The underlying pathogenesis of the aforementioned disorders is at least partially related to three well-established driver mutations resulting in constitutive activation of key cellular signaling pathways. A single-point mutation — Janus kinase 2 ( JAK2V617F) —was first discovered in 2005 when it was identified in more than 95% of patients with PV. It was also shown to be present in 50% of patients with PMF and ET. This activating mutation is localized to the pseudokinase domain of JAK2 , which normally inhibits the kinase activity of the molecule. However, the JAK2V617F mutation renders the inhibitory domain inactive, resulting in constitutive activation of the JAK/signal transducer and activator of transcription (STAT, JAK/STAT pathway) ( Fig. 5.4 ). JAK2 exon 12 mutations are present in the majority of patients with PV who do not have the JAK2V617F mutation (2% to 3% of total). The JAK2V617F mutation arises in a multipotent hematopoietic progenitor and is present in all myeloid lineages. It often undergoes transition from heterozygosity to homozygosity owing to mitotic recombination. Mutations in the myeloproliferative leukemia virus oncogene (MPL) , which encodes the thrombopoietin (TPO) receptor located in exon 10 are also associated with MPNs. Specifically, myeloproliferative leukemia (MPL) mutations are restricted to cases of ET (4%) and PMF (8%). Most recently, in 2013, mutations in the calreticulin ( CALR) gene were discovered in a majority of JAK2- and MPL- negative ET and PMF patients. CALR mutations are seen in 25% to 30% of patients with MF and 15% to 20% of patients with ET. Under physiologic conditions, CALR is a chaperone protein in the endoplasmic reticulum (ER); it assists in protein folding and is also involved in calcium homeostasis. More than 50 mutations have been discovered in exon 9 of the CALR gene; two variants, type I and type II, account for 80% of patients (type 1-52 base pair deletion is more often associated with PMF and type 2-5 base pair insertion has a slightly poorer prognosis). All of these mutations result in a gain-of-function protein, allowing for binding directly to MPL independently of thrombopoietin or other cytokine signals. The frameshift mutations seen in CALR result in excessive positive charges in the C-terminus of the mutant protein, allowing for strong binding to MPL and its consequent activation. Aberrant CALR function is thus dependent on its interaction with MPL; functional JAK2 is also a requirement, as in JAK2-deficient cell lines, CALR-dependent activation of MPL did not occur. Mutated CALR binds almost exclusively to MPL; it does not activate the EPO receptor and only weakly activates the granulocyte colony-stimulating factor receptor (GC-SFR), thus inducing a megakaryocytic MPN phenotype. Patients without any of these three driver mutations are known to have triple-negative disease, which tends to carry a worse prognosis. These so-called driver mutations are not necessarily disease-initiating events in MPNs and are not the only molecular abnormalities seen in these patients. Many additional common mutations have been described in patients with MPNs at low frequencies. These mutations are not unique to MPNs and can also be seen in other myeloid disorders, such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) ( Table 5.1 ), and can carry prognostic significance.
Case 1
A 59-year-old man referred to hematology after a routine blood cell count shows hemoglobin of 19.3g/dL and a hematocrit of 59%. Patient reports to be asymptomatic. Initial testing demonstrated EPO level of 3 and JAK2V617F allele burden of 57%.
Case 2
A 42-year-old woman presented with 5 days of abdominal pain, nausea, and vomiting. Laboratory testing showed elevated liver function values. A blood cell count showed hemoglobin of 17 with hematocrit of 53%. Abdominal imaging demonstrated portal vein thrombosis. JAK2V617F allele burden was 43%.
| Gene | Location | Mutation | Frequency |
|---|---|---|---|
| Janus kinase 2 ( JAK2 ) | 9p24 exon 14 9p24 exon 12 | JAK2 V617F | 65%Infrequent |
| Calreticulin ( CALR ) | 19p13.2 exon 9 | Type 1 (52 bp del) and type 2(5 bp in) | 25% |
| Ten-eleven translocation 2 ( TET2 ) | 4q24 | 17% | |
| Serine/arginine-rich splicing factor 2 ( SRSF2 ) | 17q25.1 | 17% | |
| U2 small nuclear RNA auxiliary factor 1 ( U2AF1 ) | 21q22.3 | 16% | |
| DNA methyltransferase 3 alpha ( DNMT3a ) | 2p23 | 15% | |
| Enhancer of zeste homolog 2 ( EZH2 ) | 7q36.1 | 13% | |
| Myeloproliferative leukemia ( MPL ) virus | 1p34 exon 10 | MPL W515L/K | 5%–10% |
| Casitas B-lineage lymphoma ( CBL ) | 11q23.3 | Exons 8 and 9 | 6% |
| Splicing factor 3B unit 1 ( SF3B1 ) | 2q33.1 | 6% | |
| Isocitrate dehydrogenase ( IDH1 and 2 ) | 2q33.3/15q26.1 | 4% | |
| Lymphocyte-specific adapter protein ( LNK ) | 12q24 | Infrequent | |
| Ikaros family zinc finger 1 ( IKZF1 ) | 7p12 | Infrequent | |
| Additional sex Combs-like 1 ( ASXL1 ) | 20q11.1 | Infrequent |
Polycythemia Vera
PV is characterized primarily by increased red cell mass, resulting in elevated hematocrit; leukocytosis, thrombocytosis, and splenomegaly are commonly present as well. The incidence of PV is reported to be 2.8 per 100,000 persons per year. Slightly more men than women develop PV, with a male-to-female ratio of ∼1:2.1. Although the average age at diagnosis is 60 years, PV can present at any age, including in children. Interestingly, females comprise 75% of patients who are younger than 40 years at diagnosis. These young patients have a much higher frequency of presentation with unique sites of thrombosis such as in the splenic vein (splanchnic thrombosis). The median survival for patients with PV varies in different studies but is shorter than age-matched control. Patients can present with a thrombotic event at diagnosis that prompts a workup showing erythrocytosis. Some patients who present with portal or hepatic vein thrombosis can have normal hemoglobin values. They may have splenomegaly or leukocytosis, which suggests a possibility of MPN and prompts JAK2 mutational testing. Other patients undergo a workup because of a presentation with systemic symptoms typical of PV, such as pruritus or visual disturbances. It is not unusual for erythrocytosis to be picked up on a routine visit in an asymptomatic patient. Typical clinical manifestations of PV are listed in Box 5.1 .
- •
General complaints: headaches, dizziness, excessive sweating, visual disturbances, weight loss
- •
Pruritus
- •
Thrombosis
- 1.
Arterial:stroke, myocardial infarction, transit ischemic attack
- 2.
Venous: deep venous thrombosis, pulmonary embolus, peripheral vascular occlusions
- 3.
Unusual anatomic sites: splanchnic veins, cerebral sinus veins, vena cava, intraventricular
- 4.
Erythromelalgia
- 1.
- •
Hemorrhagic event
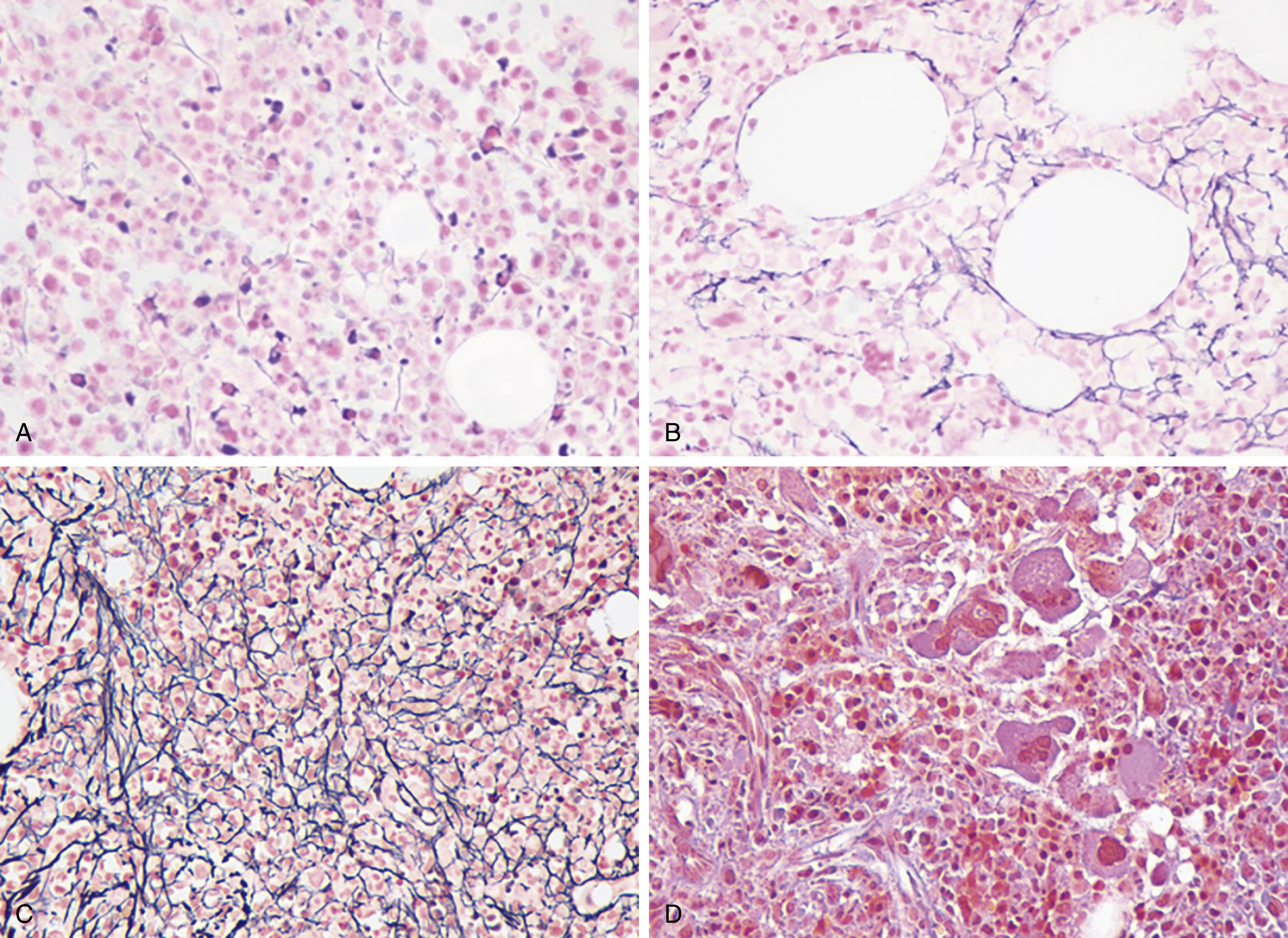
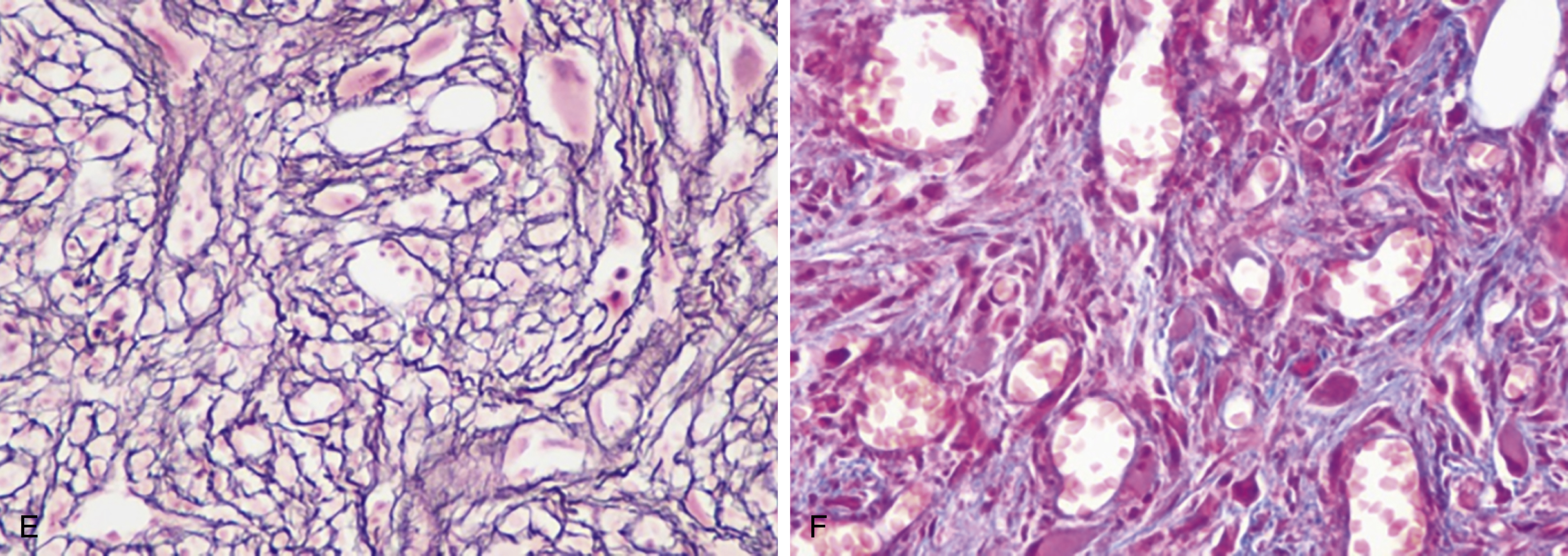
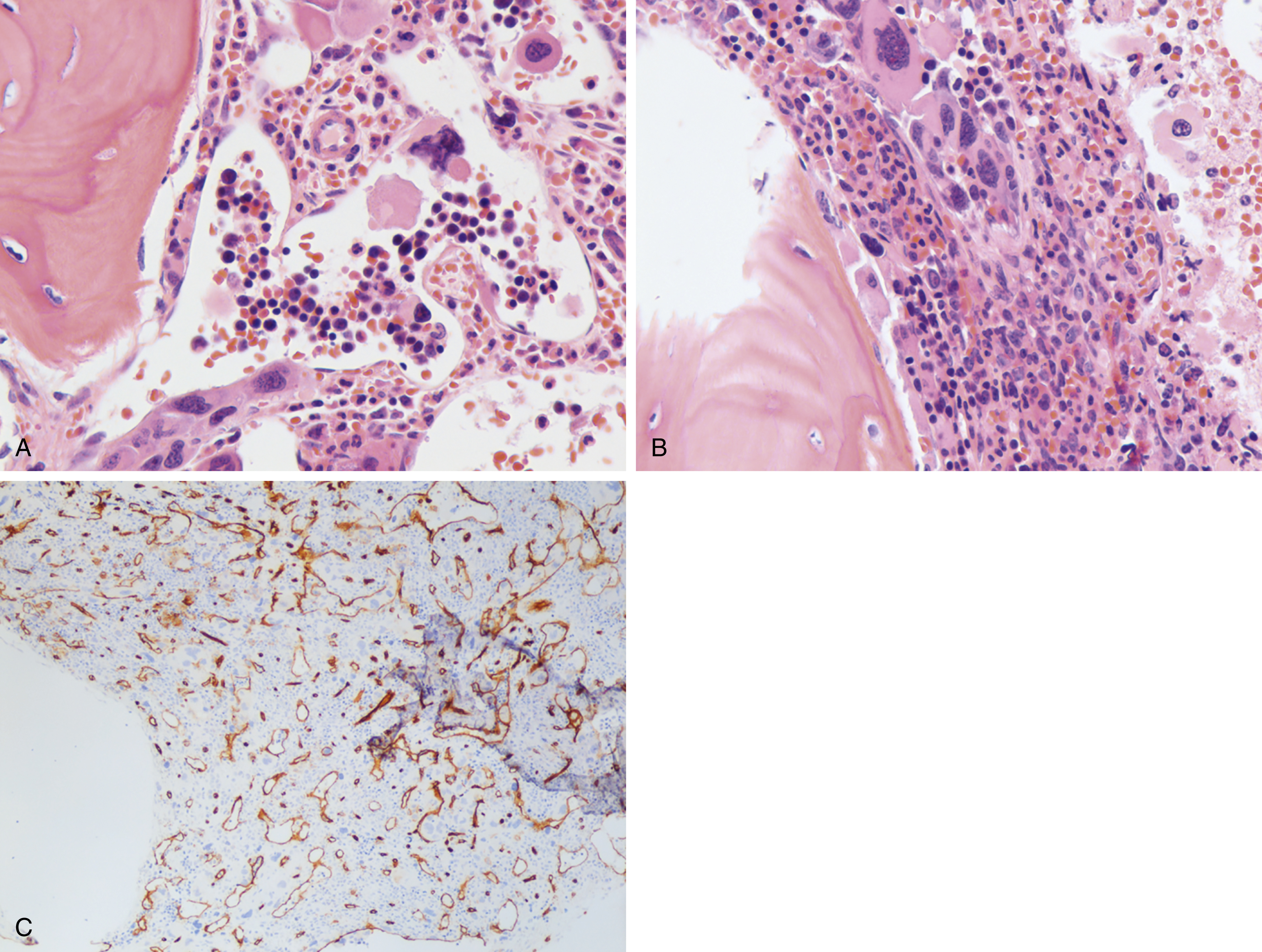
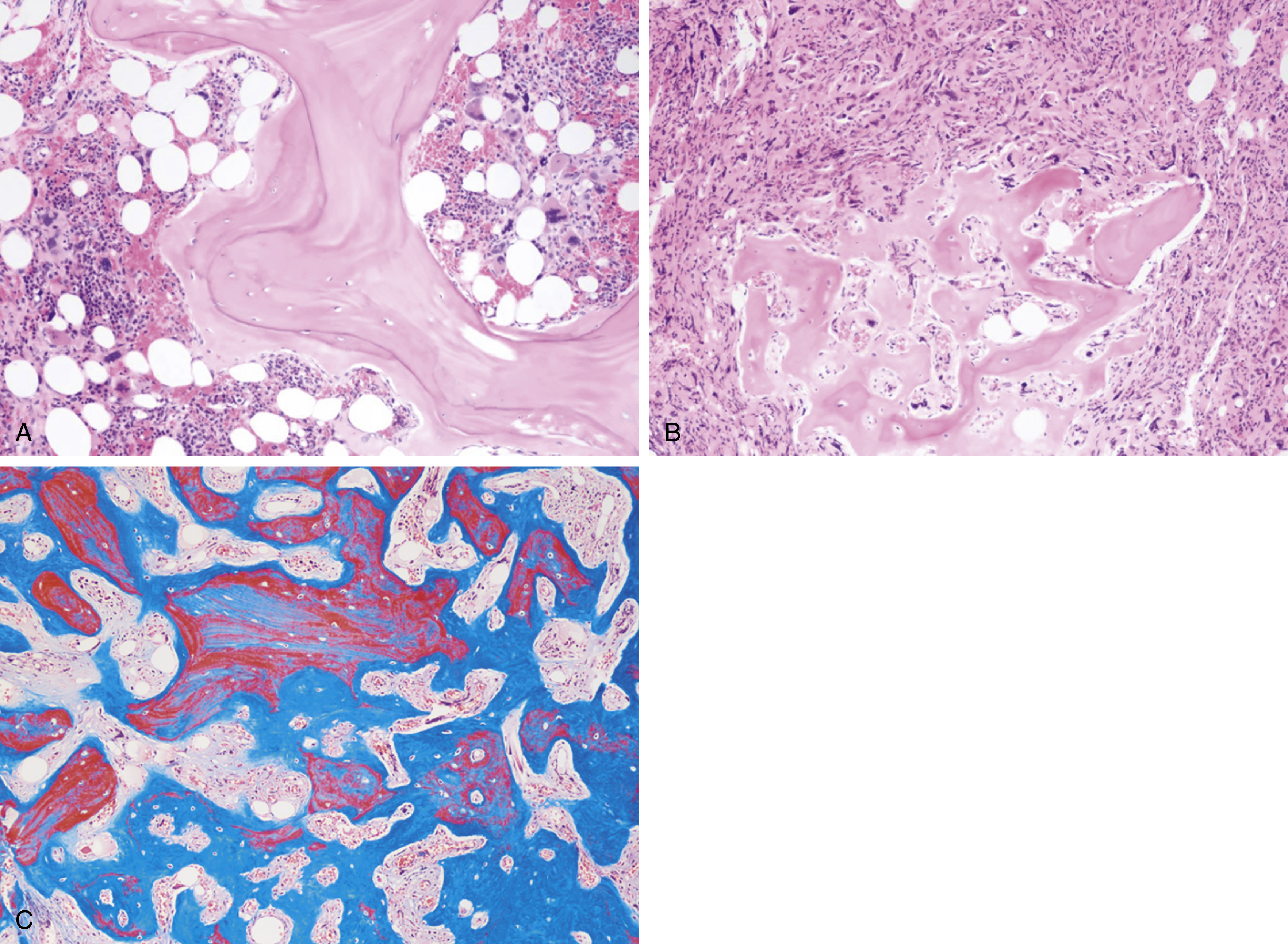
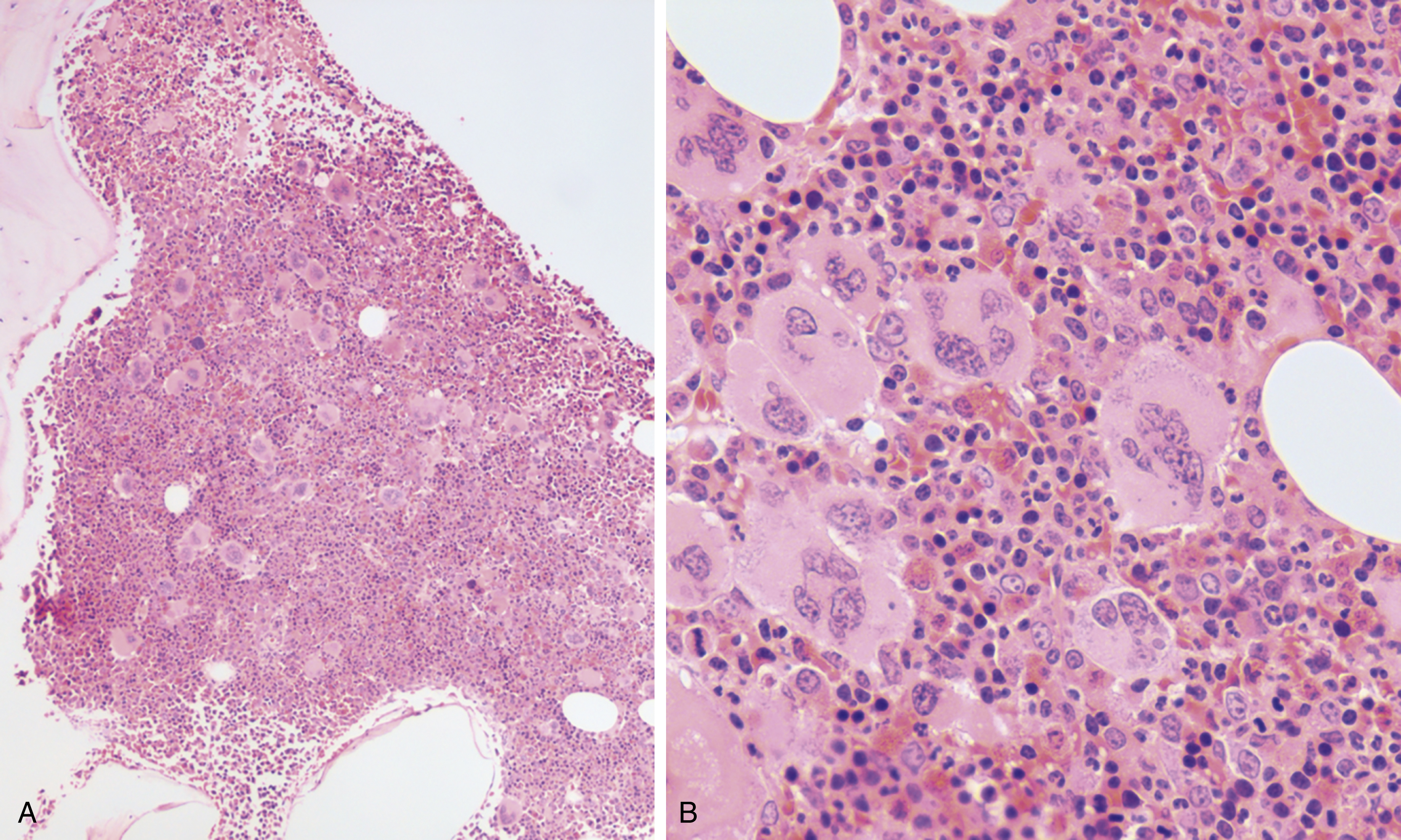
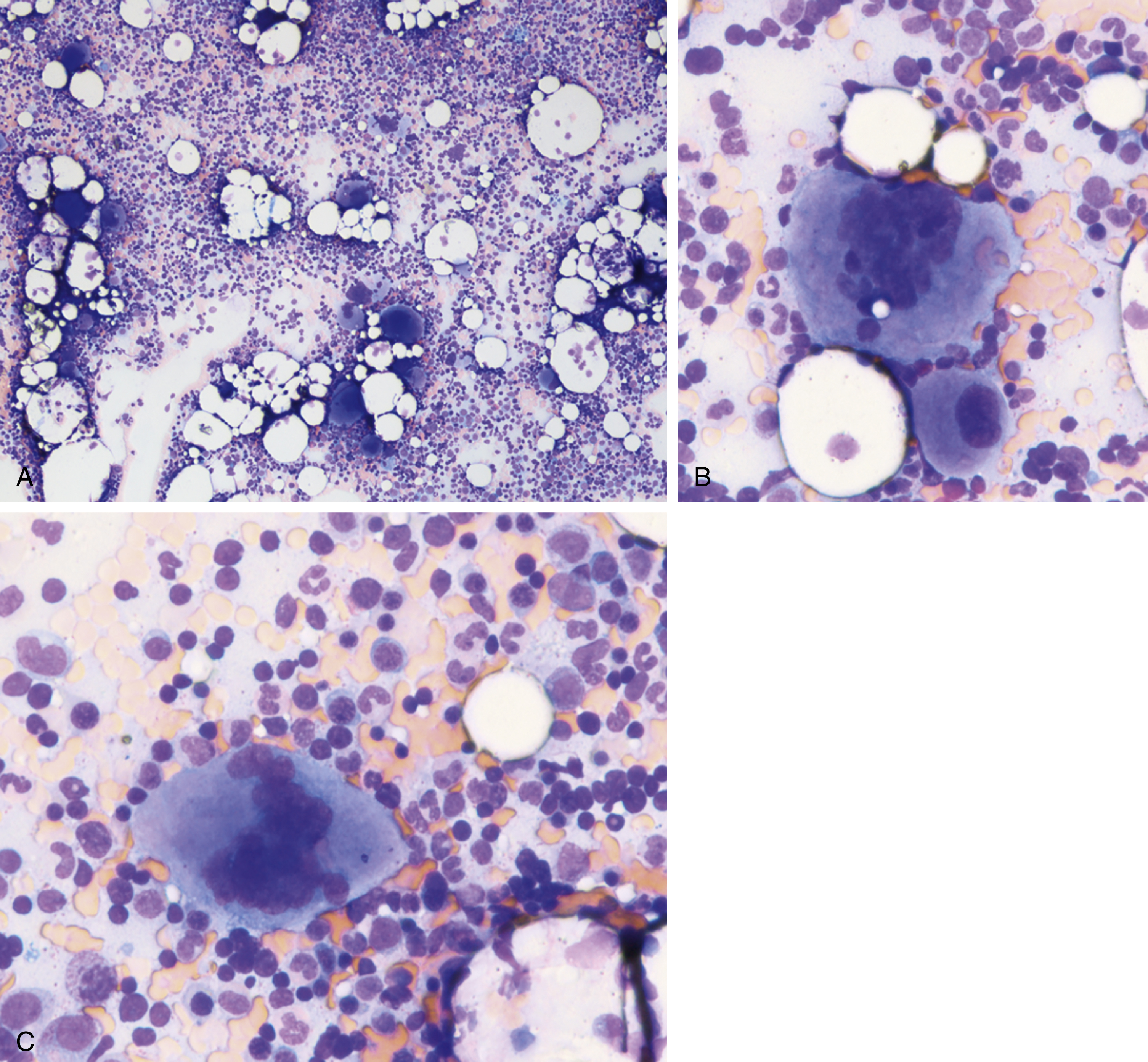
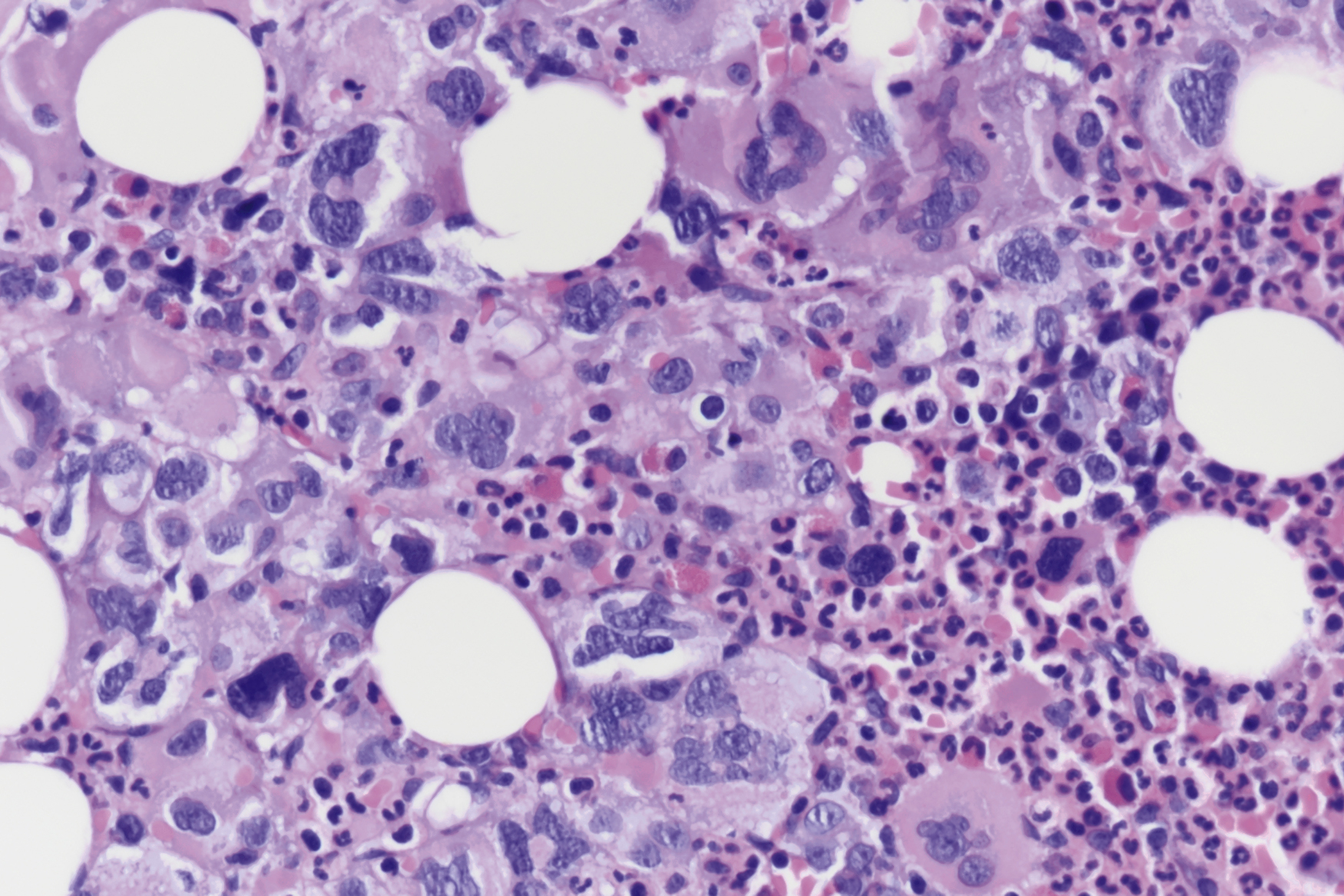
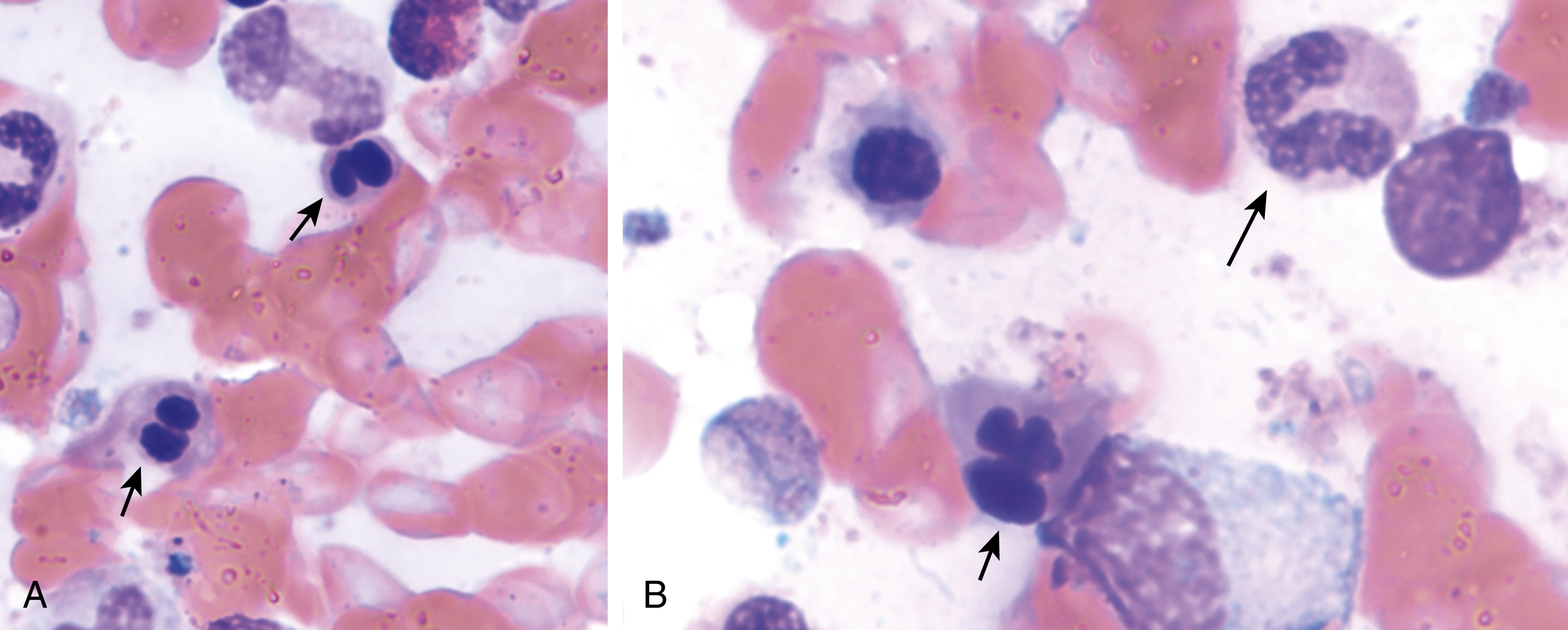
The most recent WHO diagnostic criteria are presented in Box 5.2 . Whereas BM biopsy is technically required for the diagnosis of PV in a select group of patients with extreme erythrocytosis, JAK2 mutations, and low erythropoietin (EPO) levels, the diagnosis of PV leaves little ambiguity. BM biopsy and aspirate in patients with PV are typically hypercellular and display trilineage hyperplasia with normal cellular elements ( Figs. 5.5, 5.6, 5.7 ). Iron stores are usually depleted because most patients are severely iron deficient. BM fibrosis may be observed even at the polycythemia stage as demonstrated by increased BM reticulin. It does not necessarily indicate a pending transformation to post-PV MF if the clinical picture is still consistent with the polycythemia stage. A major complication of PV is transformation to post-PV MF and AML. The risk of transformation to post-PV MF is believed to be less than 10%-15% at 20 years. Clinically, patients develop worsening splenomegaly, cytopenias, and increasing systemic symptoms. A peripheral smear demonstrates teardrop RBC morphology and a leukoerythroblastic picture. Diagnostic criteria for post-PV MF defined by the International Working Group for Myelofibrosis Research and Treatment are listed in Box 5.3 . Pathologically the BM is hyperplastic with increases in all lineages, megakaryocytic pleomorphism, and clustering and increased fibrosis ( Fig. 5.8 ). Once transformation to post-PV MF occurs, the prognosis and likelihood of survival significantly worsen. MDS-like transformation can also occur ( Fig. 5.9 ).
Case 3
A 35-year-old woman is found to have thrombocytosis of 852,000. The rest of the complete blood cell count (CBC) is unremarkable. She feels well and has no history of thrombosis or hemorrhagic episodes. There is no evidence of iron deficiency. Molecular testing demonstrates a JAK2V617F mutation with allele burden of 8%.
Major Criteria
- 1.
Hemoglobin
- •
>16.5 g/dL men
- •
>16 g/dL women or
- •
Hematocrit
- •
>49% men
- •
>48% women or
- •
- •
Red cell mass >20% above mean
- •
- 2.
Bone marrow biopsy showing hypercellularity for age with trilineage growth, including prominent erythroid, megakaryocytic, and granulocytic proliferation with pleomorphic mature megakaryocytes
- 3.
Presence of JAK2V617F or JAK2 exon 12 mutation
Minor Criteria
- •
Subnormal serum erythropoietin level
- •
Diagnosis of polycythemia vera requires either all three major criteria or the first two major criteria and the minor
Required Criteria
- 1.
Previous diagnosis of PV as defined by WHO criteria
- 2.
Bone marrow fibrosis grade 2–3 (on 2–3 scale) or grade 3–4 (on 0–4 scale)
Additional Criteria (two are required)
- 1.
Anemia or sustained loss of requirement for either phlebotomy (in the absence of cytoreductive therapy) or cytoreductive treatment for erythrocytosis
- 2.
A leukoerythroblastic peripheral blood picture
- 3.
Increasing splenomegaly, defined as either an increase in palpable splenomegaly of ≥5 cm or the appearance of a newly palpable splenomegaly
- 4.
Development of ≥1 of 3 constitutional symptoms: 10% weight loss in 6 months, night sweats, unexplained fever >37.5°C
Differential Diagnosis
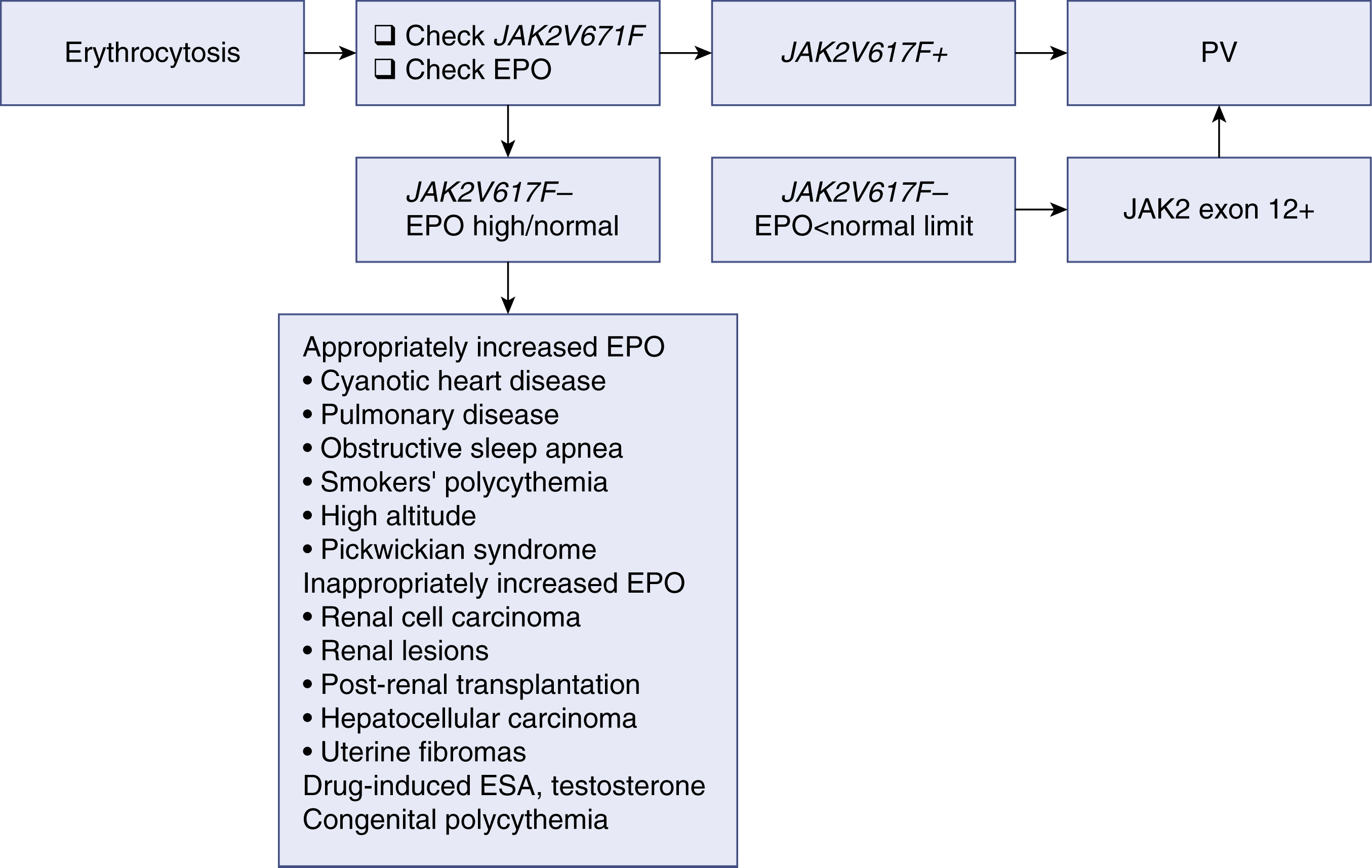
Erythrocytosis may develop from either primary or secondary causes. Primary erythrocytosis is the result of intrinsic abnormalities of hematopoietic progenitors that lead to constitutive overproduction of red cells and appropriately low EPO levels. Secondary erythrocytosis can result from increased EPO production that then acts on normal progenitor cells to increase red cell production. Polycythemia vera is considered a primary polycythemic condition; an algorithm for the diagnostic workup of erythrocytosis is shown in Fig. 5.10 . Erythrocytosis from causes other than PV can be both acquired and inherited, and a full history and physical examination help differentiate among different causes.
Essential Thrombocythemia
Essential thrombocythemia may present as an incidental finding noted on CBC or symptomatically. Even when encountering a symptomatic presentation, a review of previous CBCs often reveals a slowly increasing platelet count before presentation. Symptomatic patients may present with arterial or venous thrombotic events, including deep vein thrombosis, pulmonary embolism, splanchnic thrombosis, cavernous sinus thrombosis, ischemic stroke, or myocardial infarction. Neurologic symptoms may occur (headaches, migraines, and paresthesias of extremities) and visual disturbances are often reported. Transient ischemic attacks, which are not uncommon, can involve both posterior and anterior circulation. Microvascular vessel insufficiency in the toes and fingers can occur and present with pain in the digits. Erythromelalgia and frank gangrene can also occur. Alternatively, patients with platelet count elevations above 1 million/μL may show evidence of acquired von Willebrand (vW) disease owing to the loss of high-molecular-weight vW multimers by the excess platelet mass. Typically, such patients report a history of mucocutaneous bleeding.
The median age at diagnosis in the United States is 68 years, but ET is also found in pediatric populations. The incidence of ET is approximately 1 per 100,000 people per year. Owing to the low mortality rate with near-normal life expectancy, the prevalence is much greater than the incidence. The most common complication is thrombosis. Thrombosis-free survival rates exceed 90% in all except the highest risk stratum ( Box 5.4 ; Table 5.2 ).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


