Myeloproliferative disorders and the serum hyperviscosity syndrome can rapidly manifest with emergent presentations. Hyperviscosity occurs from pathologic elevations of either the cellular or acellular (protein) fractions of the circulating blood. Classic hyperviscosity syndrome presents with the triad of bleeding diathesis, visual disturbances, and focal neurologic signs. Emergency medicine providers should be aware of these conditions and be prepared to rapidly initiate supportive and early definitive management, including plasma exchange and apharesis. Early consultation with a hematologist is essential to managing these complex patients.
One of the most striking complications in hematologic disease is the development of acute blood hyperviscosity. Classically, hyperviscosity presents with the triad of bleeding, visual disturbances, and focal neurologic signs. Hyperviscosity occurs from pathologic elevation of either the cellular or acellular (protein) fractions of the circulating blood. In cellular fractions, significant elevation of any of the three primary blood cell lines may lead to clinical manifestations: erythrocytosis (red blood cells [RBC]), leukocytosis (white blood cells [WBC]) and thrombocytosis (platelets) ( Figs. 1 and 2 ). The term “hyperviscosity syndrome” (HVS) is best reserved for pathologic increases in circulating serum proteins, which also manifest with emergency signs and symptoms. Although the underlying disease processes vary, general management measures for any of these conditions include aggressive supportive resuscitation, rapid reduction of the offending biologic substance, and definitive chemotherapeutic management of the underlying hematologic condition. Emergency medicine providers should be aware of these conditions and be prepared to rapidly initiate supportive and early definitive management. This article is separated into four sections. The first section reviews classic HVS related to hyperproteinemia. The remainder of the article discusses the three basic lines of myeloproliferative disorders.
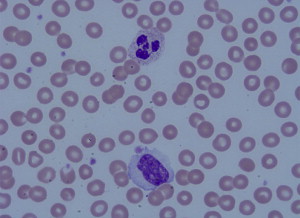
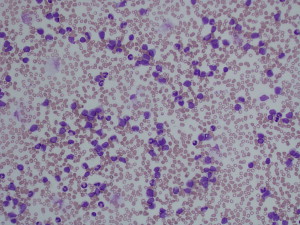
HVS related to hyperproteinemia
Waldenstrom macroglobulinemia (WM) is the most common cause of HVS. However, WM is a fairly rare disease with an incidence of only three per million persons annually (accounting for 1% to 2% of hematologic cancers). The mean age at onset of WM is about 65 years with a slight predominance in men. More than 30% all WM patients may develop HVS at some point. Under the World Health Organization (WHO) classification scheme, WM is classified separately from the myelomas.
Myelomas manifest HVS less frequently than WM but they are still the second leading cause of HVS. IgA myeloma is responsible for approximately 25% of cases of HVS followed by IgG myelomas at less than 5% of cases. Light chain disease is also a commonly reported cause of HVS. Polyclonal gammopathies from chronic infections or inflammatory disease may on occasion yield sufficiently high protein levels to create HVS, and usually respond to the same general treatment. Sjögren syndrome, rheumatoid arthritis, systemic lupus erythematosus, cryoglobinemia, diabetes mellitus, and even HIV, have been associated with HVS.
Pathophysiology
Viscosity is formally defined as the internal frictional resistance of fluid to flow, or in simple terms, the “thickness” of the fluid. Water has a lower viscosity than syrup; it therefore travels faster and easier, especially through small passageways. Similarly, hyperviscous blood creates sludge and stasis in the capillary beds resulting in clinical manifestations at the tissue and circulatory level. Viscosity derives from physical and chemical properties, most notably the concentration, the size, and the shape of the molecules. Centipoise (cp) is the standard unit for measuring dynamic fluid viscosity, named in honor of the French physiologist Jean Louis Marie Poiseuille (who also described Poiseuille’s Law). Water has a viscosity of 1.00 cp at 20°C The large protein IgM pentameters in WM are highly viscous but HVS can also be seen in kappa light chain disease, which tends to form unstable, asymmetric, highly polymerized circulating aggregates. The clinical manifestations of HVS appear when the serum viscosity relative to water is greater than about 3 cp; at higher levels of 4cp and 5cp the prevalence of HVS rises to 67% and 75%, respectively. The underlying physiology of the patient creates significant variability from one patient to another, but for a given patient symptoms will manifest at about the same level of viscosity over time.
Clinical Presentation
HVS should be considered in at least three different Emergency Department (ED) scenarios: the patient presenting with the classic triad of HVS; the ED patient who has a previously established immunoglobulin-producing hematologic diagnosis; and critically ill patients with certain laboratory clues pointing to a concurrent underlying gammopathy.
The classic presentation triad for HVS consists of bleeding, visual disturbances, and focal neurologic signs. However, a variety of end organ damage can be observed. The bleeding typically arises from oozing mucosal surfaces including epistaxis, bleeding gums and gastrointestinal hemorrhage due to a supposed mechanism of impaired platelet function. Retinopathy caused by thrombosis, microhemorrhage, exudates, and papilledema results in the common visual derangements. These findings can be directly observed under fundoscopy with the classic picture of “sausage link” or “boxcar” venous engorgement of the retinal veins. Neurologic complications may present with headache, generalized stupor, and coma, or focal findings including vertigo, hearing impairment, seizures, and stroke syndromes. Not in the diagnostic triad, but often more hazardous, are the cardiopulmonary complications of HVS. Among other mechanisms, the effectively expanded plasma volume seen in HVS may drive demand beyond cardiac reserve resulting in high output cardiac failure, valvular dysfunction, or myocardial infarction.
Physicians should consider the diagnosis of HVS if a patient presents with unexplained neurologic symptoms such as visual change or headache in the setting of a concomitant immunoglobulin-producing hematologic condition. HVS is often the presenting characteristic of dysproteinemias. Renal failure has been reported with HVS but is generally more common in myelomas. One mechanism of renal failure in HVS may be that the glomeruli are exposed to a relative hypoperfused “pre-renal” state. Judicious hydration and avoiding use of diuretics is important to prevent further morbidity. Myeloma patients are notoriously susceptible to radiologic contrast nephropathy, so great caution should be exercised in its administration. HVS is also one cause of symmetric peripheral gangrene of the extremities.
Laboratory evaluation should include a complete blood cell count, full serum chemistries, and coagulation profiles. Significant proteinuria on routine urinalysis and an elevated albumin-protein gap (albumin − protein difference) suggests an underlying gammopathy. Occasionally, the laboratory will report the presence of rouleaux formation on the peripheral blood smear or that they are unable to complete tests because the viscous serum has clogged their analytical instruments. Paraneoplastic signs of multiple myeloma include hypercalcemia and hyponatremia. Confirmatory serum and urine protein electrophoresis with the characteristic monoclonal spike on serum electrophoresis confirms an existing gammopathy; this test can be arranged in conjunction with the admitting physician.
Diagnosis
HVS is a clinical diagnosis made on a combination of the symptoms discussed earlier in conjunction with a viscosity of 4 cp or greater as measured by a viscosimeter (normal viscosity is 1.4–1.8 cp). Controversy exists on whether plasma viscosity or whole blood viscosity is better, but measurement of whole blood viscosity is probably superior to that of serum or plasma viscosity alone. Laboratories report whole blood viscosity as comparable to a sample with a certain hematocrit (a viscosity comparable to that of a patient with a hematocrit of 70%). A relative whole blood viscosity of 55% or greater increases the risk of HVS.
Management
Physicians should base treatment decisions primarily on the severity of signs and symptoms rather than the measured degree of hyperviscosity. HVS therapy can be viewed in 4 phases: supportive therapy, plasma exchange, plasmapheresis, and chemotherapy for the underlying hematologic condition. Specific chemotherapy is generally applied after the ED course by hematology/oncology consultants and is therefore beyond the scope of this article.
Supportive Therapy
Paradoxically, these patients may be dehydrated yet present with high output cardiac failure. Judicious fluid resuscitation is usually necessary and diuretics should be avoided. Electrolyte derangements should be corrected with the foreknowledge that sodium derangements are often spurious. Antibiotics should be administered if any suspicion of infection exists. Although fluid compartment shifts may result in a measured anemia, these patients generally have a normal or supernormal red blood cell mass. Because packed red blood cell transfusions increase blood viscosity and thereby accelerate the HVS, they should be withheld until the viscosity can be measured and lowered appropriately.
Plasma Exchange
In the ED, one temporizing measure in a patient with severe HVS is to perform an emergency plasma exchange. The procedure involves phlebotomizing between 1 and 2 units of blood from the patient while simultaneously replacing the volume with normal saline. This therapy is of proven benefit and some experts believe it is more effective than plasmapheresis in decreasing serum viscosity in patients with WM. However, aggressive plasma exchange can deplete platelets, clotting factors, and albumin. Emergency plasma exchange is indicated in the presence of severe neurologic findings (seizure or coma). Measuring quantitative immunoglobulins before and after plasma exchange can help guide further hospital therapy but does not influence the ED course. IgA and IgG molecules have a higher volume of distribution (compared with the IgM of WM) and therefore plasma exchange may need to be performed more aggressively in these myelomas.
Plasmapheresis
Plasmapheresis is the definitive treatment for HVS. The ED physician should establish large-bore central venous access while awaiting the plasmapheresis team, preferably with a dialysis catheter. As noted, IgG- or IgA-related HVS will require longer and more frequent plasmapheresis treatments. The IgM found in WM is mostly intravascular and therefore is cleared more readily. The major complication of plasmapheresis is hypocalcemia related to citrate binding from automated plasmapheresis systems.
Erythrocytosis and polycythemia vera
Erythrocytosis results from an increase in the red cell mass with concomitant increase in RBC number, red cell count, and hematocrit. This finding may be generally attributed to hemoconcentration given the many cases of dehydration, hypovolemia and other relative low-volume states encountered in the ED. However, the critically ill patient presenting with an acute increase in hematocrit or in association with increases in other blood cell components (ie, platelets, leukocytes) requires consideration of a more aggressive diagnostic workup.
The incidence of polycythemia vera (PCV) is about 2.6 cases per 100,000 persons, and is highest among Ashkenazi Jews. PCV is associated with a point mutation of an auto-inhibitory Janus kinase 2 (JAK2) protein kinase domain. The activation of this domain results in erythropoeisis losing its dependence on erythropoietin signaling and becoming virtually autonomous. Recent studies have identified several additional mutations within the JAK2 gene site that are now considered essential to the diagnosis of PCV.
Pathophysiology
Hematopoiesis exists in a homeostatic balance between the body’s requirements for particular blood cell lines and loss or destruction of those cells. In the red cell line this balance is maintained by a feedback mechanism primarily involving the hormone erythropoietin. Erythropoietin is primarily produced in the renal cortex, accounting for 90% of this circulating protein. Secondary sites of production consist of liver, spleen, lung, testis, brain, and erythroprogenitor cells. Erythropoietin stimulation results in the production of 2 × 10 11 red blood cells per day. All blood cell lines arise from a common hematopoetic stem cell. These stem cells begin their initial differentiation onto erythrocyte progenitors when stimulated by one of several cytokine factors.
Erythrocytosis is the proper term for a state of increase in circulating red cells. Specifically, PCV possesses these additional features and has potential for progression to advanced disease. Erythrocytosis is initially identified by the increased in hematocrit and red blood cell count, but it is important to remember that these indices are dependent on red blood cell mass and plasma volume. Thus, a relative erythrocytosis occurs in cases of dehydration and low volume due to hemoconcentration. Correction in these cases can be accomplished by volume replacement and the condition may be short-lived.
An absolute erythrocytosis is associated with an increase in red cell production and mass. These patients are euvolemic with an increased hematocrit. Absolute erythrocytosis has been classified as either primary or secondary. Secondary erythrocytosis consists of congenital and acquired disorders. Congenital conditions are related to high oxygen affinity hemoglobinopathies and gene mutations (eg, 2,3-diphosphoglycerate deficiency). Acquired conditions are associated with erythropoietin-mediated erythrocyte production. This erythropoietin response may be hypoxia in patients living in high altitudes, those with chronic lung disease, or from paraneoplastic effects.
Diagnosis
The diagnosis begins with determining the red cell mass/body surface area in patients presenting with a persistently increased hematocrit in the absence of secondary causes of erythrocytosis. The Polycythemia Vera Study Group and the World Health Organization (WHO) have issued criteria for the diagnosis of PCV ( Box 1 ). New, proposed WHO criteria incorporate the presence of erythrocytosis or increased red cell mass and the presence of a known PCV molecular lesion.
Major Criteria
Hemoglobin >18.5 g/dL in men
Hemoglobin >16.5 g/dL in women
Or increase red cell volume
Presence of JAK2V617F or JAK2 exon 12 mutation
Minor Criteria
PCV bone marrow changes
Low serum erythropoietin level
Endogenous erythroid colony formation in vitro
Data from Ref.
Treatment
Phlebotomy and low-dose aspirin are the mainstays of PCV treatment. The current recommendations are phlebotomy to a hematocrit of ≤45% in men and ≤42% in women. However, no evidence exists for this goal of phlebotomy other than correlation between increased risk of thrombosis and a hematocrit >45% in these patients. Low-dose aspirin (either 81 or 100 mg daily) is recommended unless there are specific contraindications. More recent studies support initiation of aspirin therapy even in those patients with a previous history of gastric bleeding in the setting of concurrent use of a proton pump inhibitor. Further treatment is individualized based on the patient’s risk for thrombotic complications. A history of thrombosis, age more than 60 years, and associated thrombocytosis are indications for more aggressive treatment.
The next step in the management of higher risk patients is the addition of cytoreductive therapy. Options include hydroxyurea, chlorambucil, or intravenous radioactive phosphorus-32. Recombinant interferon alpha is another option in myelosuppressive management. Secondary erythrocytosis may be managed initially with phlebotomy. However, definitive management will require the primary cause to be addressed. Malignancy, hypoxia, and extrinsic sources of erythropoietin should be addressed and managed accordingly. These patients are also at risk for neurologic sequelae.
Erythrocytosis and polycythemia vera
Erythrocytosis results from an increase in the red cell mass with concomitant increase in RBC number, red cell count, and hematocrit. This finding may be generally attributed to hemoconcentration given the many cases of dehydration, hypovolemia and other relative low-volume states encountered in the ED. However, the critically ill patient presenting with an acute increase in hematocrit or in association with increases in other blood cell components (ie, platelets, leukocytes) requires consideration of a more aggressive diagnostic workup.
The incidence of polycythemia vera (PCV) is about 2.6 cases per 100,000 persons, and is highest among Ashkenazi Jews. PCV is associated with a point mutation of an auto-inhibitory Janus kinase 2 (JAK2) protein kinase domain. The activation of this domain results in erythropoeisis losing its dependence on erythropoietin signaling and becoming virtually autonomous. Recent studies have identified several additional mutations within the JAK2 gene site that are now considered essential to the diagnosis of PCV.
Pathophysiology
Hematopoiesis exists in a homeostatic balance between the body’s requirements for particular blood cell lines and loss or destruction of those cells. In the red cell line this balance is maintained by a feedback mechanism primarily involving the hormone erythropoietin. Erythropoietin is primarily produced in the renal cortex, accounting for 90% of this circulating protein. Secondary sites of production consist of liver, spleen, lung, testis, brain, and erythroprogenitor cells. Erythropoietin stimulation results in the production of 2 × 10 11 red blood cells per day. All blood cell lines arise from a common hematopoetic stem cell. These stem cells begin their initial differentiation onto erythrocyte progenitors when stimulated by one of several cytokine factors.
Erythrocytosis is the proper term for a state of increase in circulating red cells. Specifically, PCV possesses these additional features and has potential for progression to advanced disease. Erythrocytosis is initially identified by the increased in hematocrit and red blood cell count, but it is important to remember that these indices are dependent on red blood cell mass and plasma volume. Thus, a relative erythrocytosis occurs in cases of dehydration and low volume due to hemoconcentration. Correction in these cases can be accomplished by volume replacement and the condition may be short-lived.
An absolute erythrocytosis is associated with an increase in red cell production and mass. These patients are euvolemic with an increased hematocrit. Absolute erythrocytosis has been classified as either primary or secondary. Secondary erythrocytosis consists of congenital and acquired disorders. Congenital conditions are related to high oxygen affinity hemoglobinopathies and gene mutations (eg, 2,3-diphosphoglycerate deficiency). Acquired conditions are associated with erythropoietin-mediated erythrocyte production. This erythropoietin response may be hypoxia in patients living in high altitudes, those with chronic lung disease, or from paraneoplastic effects.
Diagnosis
The diagnosis begins with determining the red cell mass/body surface area in patients presenting with a persistently increased hematocrit in the absence of secondary causes of erythrocytosis. The Polycythemia Vera Study Group and the World Health Organization (WHO) have issued criteria for the diagnosis of PCV ( Box 1 ). New, proposed WHO criteria incorporate the presence of erythrocytosis or increased red cell mass and the presence of a known PCV molecular lesion.
Major Criteria
Hemoglobin >18.5 g/dL in men
Hemoglobin >16.5 g/dL in women
Or increase red cell volume
Presence of JAK2V617F or JAK2 exon 12 mutation
Minor Criteria
PCV bone marrow changes
Low serum erythropoietin level
Endogenous erythroid colony formation in vitro
Data from Ref.
Treatment
Phlebotomy and low-dose aspirin are the mainstays of PCV treatment. The current recommendations are phlebotomy to a hematocrit of ≤45% in men and ≤42% in women. However, no evidence exists for this goal of phlebotomy other than correlation between increased risk of thrombosis and a hematocrit >45% in these patients. Low-dose aspirin (either 81 or 100 mg daily) is recommended unless there are specific contraindications. More recent studies support initiation of aspirin therapy even in those patients with a previous history of gastric bleeding in the setting of concurrent use of a proton pump inhibitor. Further treatment is individualized based on the patient’s risk for thrombotic complications. A history of thrombosis, age more than 60 years, and associated thrombocytosis are indications for more aggressive treatment.
The next step in the management of higher risk patients is the addition of cytoreductive therapy. Options include hydroxyurea, chlorambucil, or intravenous radioactive phosphorus-32. Recombinant interferon alpha is another option in myelosuppressive management. Secondary erythrocytosis may be managed initially with phlebotomy. However, definitive management will require the primary cause to be addressed. Malignancy, hypoxia, and extrinsic sources of erythropoietin should be addressed and managed accordingly. These patients are also at risk for neurologic sequelae.
Hyperleukocytosis
Leukocytosis refers to an increase in the total number of white blood cells in circulation. By definition it is an elevation greater than two standard deviations above the mean circulating white blood cell count based on the age. Hyperleukocytosis is an extreme elevation of the blast count or white blood cell count greater than 100,000/mm 3 . It occurs in malignant and nonmalignant disorders, especially acute leukemia. Hyperleukocytosis occurs more frequently in acute leukemia than in chronic leukemia, and its incidence ranges from 5% to 13% in adult acute myeloid leukemia (AML) and from 10% to 30% in adult acute lymphoblastic leukemia (ALL). Risk factors for hyperleukocytosis include age <1 year, male gender, certain subtypes of leukemia (French-American-British (FAB) Classification M4, M5), and select cytogenetic abnormalities (11q23 rearrangements and the Philadelphia chromosome).
Leukocytosis and leukostasis should be considered a medical emergency as the mortality rate may approach 40%. The management of acute hyperleukocytosis and leukostasis involves supportive measures and reducing the number of circulating leukemic blast cells. Patients may also present with tumor lysis syndrome although this is more often seen after therapy for aggressive hematological disorders such as acute leukemia. Tumor lysis syndrome is discussed in detail in a separate article in this volume.
Pathophysiology
The granulocytic and lymphocytic cell lines are routinely measured in the ED. The granulocytic series is primarily involved in phagocytic activities and is derived from a common progenitor cell located in the bone marrow that also gives rise to erythrocytes, megakaryocytes, eosinophils, basophils, and monocytes. Granulocytes are maintained in a series of developmental and storage pools. The most important is the postmitotic storage pool for neutrophils, which represents 15 to 20 times the circulating population and contains metamyelocytes, band neutrophils, and mature neutrophils. The pool can be drawn on as a ready reserve during rapid consumption of granulocytes. Cytokines and complement components release granulocytes from the marrow storage pool into the circulation, which can result in a two- to threefold increase in the granulocyte count within 4 to 5 hours. Circulating neutrophils are subdivided equally into the circulating neutrophil pool and the marginal pool. The marginal pool consists of mature cells adherent to the blood vessel walls, which can rapidly enter the circulating pool and can cause a doubling of the WBC count.
A leukemoid reaction is an excessive leukocytic response in the peripheral blood of the granulocytic cell line. The WBC exceeds 50,000/mm 3 and resembles chronic myeloid leukemia. A leukemoid reaction is characterized by a significant increase in neutrophils in the peripheral blood and a differential demonstrating a left shift. The precursors (myelocytes, metamyelocytes, promyelocytes, and myeloblasts) may occasionally be observed in severe reactions. In contrast to acute leukemia, proliferation and orderly maturation of all normal myeloid elements is observed in the bone marrow and the morphology of the myeloid elements is normal. Diagnosis of a leukemoid reaction should only be made after excluding a hematologic malignancy.
The morbidity of hyperleukocytosis results primarily from the leukostasis syndrome caused by the increased viscosity and sluggish flow of circulating leukemic blasts in tissue microvasculature. A postulated mechanism suggests an increase in blood viscosity and blockade of the microcirculation resulting in organ damage. More recent evidence suggests that interactions between leukemic blasts and the surface of endothelial cells might be responsible for the aggregation in the microcirculation. Increased production of cytokines and expression of adhesion markers such as ICAM-1, VCAM-1, and selectin by endothelial cells enables myeloblasts to recruit additional myeloblasts. This forms a vicious cycle in which increasingly more cells are trapped in the microcirculation.
Diagnosis
A unique problem with white blood cell disorders is the wide variability of normal quantitative values. Total WBC and neutrophil count in neonates younger than 1 week are physiologically higher than those in older children and adults; however, young infants less than 3 months old have smaller storage pools of neutrophils. The proportion of lymphocytes and absolute lymphocyte counts in children are higher than those in adults. Failure to recognize age-specific lymphocytosis may lead to unnecessary investigations.
Clinical Presentation
In general, leukostasis is observed in AML if the WBC is >100 × 10 9 /L and in ALL if the WBC is >400 × 10 9 /L. Leukostasis is usually associated with a high number of circulating blasts but has also been described with blast counts less than 50,000/mm 3 . Hyperleukocytosis is seen in up to 13% of patients with AML, is less common in ALL, and even less common in chronic myeloid leukemia (CML) and chronic lymphocytic leukemia (CLL). The frequency of complications is higher in AML than in ALL because the myeloblasts are larger and more adhesive than lymphoblasts. Leukostasis is present in 12% of adult patients with CML and in up to 60% of pediatric cases, but CML represents only 2% to 7% of childhood leukemias.
The manifestations of acute hyperleukocytosis are protean ( Box 2 ). Fever is common, but can be traced to definitive infection in only a small number of these patients. Blood cultures should still be obtained so that infection can be ruled out, because hyperleukocytosis can mimic several viral, bacterial, and fungal syndromes.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


