Multiple Myeloma: Current Perspectives
Quest Diagnostics Nichols Institute, 14225 Newbrook Drive, Chantilly, VA 20151, USA
Keywords
• Multiple myeloma • Cytogenetics • Fluorescence in situ hybridization • Monoclonal gammopathy • Plasma cell dyscrasia • Microarray
Introduction
Multiple myeloma (MM) is a rarely curable malignancy defined by a proliferation of monoclonal plasma cells (PCs) in the bone marrow (BM), often with an elevated serum or urine paraprotein (M protein, or monoclonal protein). It is the second most common hematologic cancer in the United States and Europe, with an incidence of 20,000 or more cases per year,1,2 representing 1% of all cancers and 2% of all cancer deaths. MM is primarily a disease of the elderly (median age at diagnosis, 67 years) with only approximately 3% of patients diagnosed before the age of 40. The cause of MM remains unknown, but associated risk factors include obesity and a family history of lymphoid hematopoietic cancers. The disease is more prevalent in men than in women and is highest in blacks and lowest in Asians.3
MM is heterogeneous at many levels, including clinical presentation, pathogenetic characteristics, response to treatment, and outcome. MM emerges through a multistep transformation from one or two asymptomatic, premalignant phases—monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM).4,5 Patients with symptomatic MM present with one or more features of CRAB, an acronym for hypercalcemia, renal insufficiency, anemia, and bone lesions.6 The International Myeloma Working Group laboratory criteria for the different stages of MM are shown in Table 1.6
MGUS, with a reported incidence of about 3% per year in people aged older than 60 years, and the more advanced premalignant stage of SMM share the same primary genetic aberrations detected in MM. They are not treated, however, because of the low likelihood of progression and the unknown effects of early intervention. The risk of progression to MM is about 1% per year for MGUS patients and about 10% per year for the first 5 years for SMM patients. Many patients remain under observation for years before transformation occurs.7,8 There is also no supporting evidence that early intervention in asymptomatic disease will improve overall survival (OS) or quality of life, lead to unexpected complications such as secondary myeloid neoplasia, or result in an earlier onset or more aggressive disease.8 MM, in contrast, with destruction of bone and suppression of bone marrow function, among other symptoms, requires immediate treatment.
The critical event in MM pathogenesis is thought to be the transformation of a proliferative “plasmablastic” cell located in the lymphoid germinal centers.9,10 The progeny of this “stem cell” migrate to the bone marrow where they mature to terminally differentiated antibody-secreting PCs. Acquired cytogenetic and molecular alterations associated with malignant transformation of PCs dysregulate the normal developmental pathway, leading to altered cell behavior. Based on the premise that hyperdiploidy or an aberrant IGH gene rearrangement leading to cyclin D dysregulation is an early, disease-defining event in the transformation process,9,11,12 Gonzalez and colleagues9 compared serial samples collected from patients in transition from MGUS to MM or at relapse and found no intraclonal diversity. These data suggest that from the precursor states of MGUS and SMM to MM, the abnormal plasma cell clone carries the same primary chromosomal aberration (IGH translocations or hyperdiploidy) and secretes the same paraprotein for the duration of the disease.8,9 Fulminant MM and its progression are typically associated with the presence of additional karyotypic aberrations, shorter therapeutic responses, and shortened OS.
Heterogeneity of disease course and outcome shows a striking relationship with the genetic alterations of the malignant PCs. Traditionally, MM has been associated with a poor prognosis and an OS of 3 to 7 years from diagnosis, although the range has varied from less than 1 year in patients with aggressive disease to greater than 10 years in patients with indolent disease. But the management of patients with MM has changed dramatically over the past decade. Emerging new therapies, such as thalidomide, lenalidomide, and bortezomib, have increased the median OS to 4 to 5 years and a 20% chance of surviving to greater than 10 years.13–15 Despite these achievements, MM is usually considered incurable. Refinement of the current genetically defined risk groups is needed to identify high-risk patients who may benefit from novel treatments from patients with predictable clinical courses who benefit from existing treatments. To address this need, the International Myeloma Working Group recently proposed consensus guidelines for the workup of patients with suspected MM using standardized diagnostic, prognostic, and response criteria.6,16–18 Currently, patient stratification is based on the known pathogenetic factors of age; tumor burden; renal function; serum levels of lactic dehydrogenase (LDH); β2-microglobulin (Sβ2M); and albumin, plasma cell labeling index, functional status, and cytogenetics (Table 2). This stratification model will soon be strengthened with the incorporation of oncogenomic diagnostic techniques including analysis of DNA copy number alteration by array comparative genomic hybridization (aCGH), single nucleotide polymorphism (SNP) microarrays, and gene expression profiling (GEP).18–20 This article reviews the conventional cytogenetics (CC), molecular cytogenetics, and genomic diagnostics of MM and highlights a few recent clinical trials that demonstrate the impact of genetic risk stratification on the treatment of this plasma cell malignancy.
Table 2 International Myeloma Working Group risk stratification guidelines for multiple myeloma
| Minimal recommendations for risk stratification | Additional studies suggested for optimal risk stratification |
| Serum albumin and β2-microglobulin assays | Conventional (metaphase) cytogenetics |
| Bone marrow examination with FISH to detect t(4;14), t(14;16), and del(17p) in PCsa,b | Labeling index |
| Lactic acid dehydrogenase levels | MRI/PET imaging |
| Immunoglobulin type | Microarrays (CGH/SNP) to determine DNA copy number alterations |
| Histology to determine plasmablastic disease | Gene expression profiling |
a Identified by plasma cell specific FISH protocols.
Historical Perspective of Cytogenetics in MM
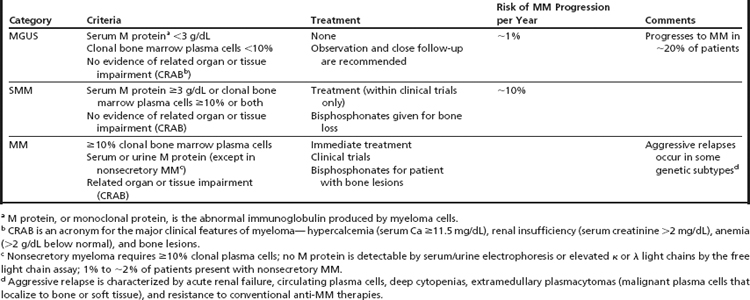
Dewald and colleagues were the first to describe the clinical utility of chromosome studies in MM.21 In their seminal paper, they showed that compared with patients with karyotypically normal bone marrow cells, patients with karyotypically abnormal bone marrow cells had reduced OS, underscoring the relevance of the plasma cell genome to disease outcome. Normal PCs are terminally differentiated cells with low proliferative activity. The presence of karyotypically normal bone marrow studies in MM patients provides clear evidence of active, normal hematopoiesis, whereas patients with abnormal karyotypes show a higher proliferative, less differentiated PC neoplasm indicative of active disease and higher tumor burden. Over the following 15 years, conventional cytogenetic studies reported an incidence of abnormal PC karyotypes in 30% to approximately 40% of newly diagnosed and 35% to approximately 60% of previously treated MM patients.22–27 Despite the relatively low abnormality rate, the data exposed two clinically significant findings: (1) conventional cytogenetic studies are a surrogate for the plasma cell proliferative index assay, with either test capable of indicating active proliferating disease in the bone marrow; and (2) prognostic differences are associated with ploidy level and with specific recurring cytogenetic aberrations (Fig. 1).
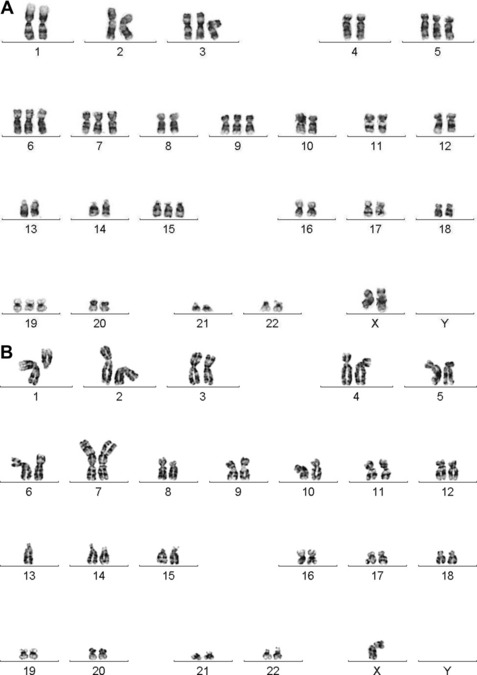
Because the low proliferative potential of terminally differentiated PCs limits detection of chromosomal aberrations by CC, which requires metaphases, interphase fluorescence in situ hybridization (I-FISH) has become an attractive technique to interrogate MM-specific chromosomal aberrations. All patients with abnormal karyotypes show known abnormalities by I-FISH, but the two assays provide slightly different information. I-FISH detects specific prognostically significant cytogenetic markers in malignant PCs, in particular, high-risk IGH translocations such as t(4;14)/IGH/FGFR3 and t(14;16)/IGH/MAF that are not obvious by CC because they are cryptic or hidden in complex karyotypes.28 However, I-FISH detects only its intended target and provides no information about additional abnormalities that may be present or may signal disease progression. Metaphase cytogenetics, however, is an excellent tool for detecting the wide range of karyotypic alterations observed in MM and, as mentioned previously, serves as a surrogate index for active proliferative disease. Thus, I-FISH is not a substitute for CC; it has clear clinical utility when CC is either noninformative or not interpretable and is a necessary adjunct to metaphase cytogenetics for detecting subtle IGH translocations. In addition, both interphase and metaphase FISH studies extend the advantage that the residual cell pellet from metaphase studies or the G-banded slides can be used to refine data gleaned from the CC study. Together, CC and FISH (both metaphase and interphase) play important and independent roles in MM stratification by providing timely and prognostically relevant information for newly diagnosed patients and by defining secondary aberrations associated with clonal evolution.
CC Methods
A comprehensive analysis includes G-banded karyotyping and metaphase FISH, when applicable. With metaphase cytogenetics alone, a laboratory can expect informative results in approximately 40% of cases, but metaphase FISH is required to show cryptic or hidden IGH fusions and deletions in approximately 10% of cases.28 Periodic follow-up cytogenetic studies should be recommended to detect clonal evolution of disease or evolving therapy-related myeloid neoplasms.
FISH Studies
Dewald and colleagues28 reported that bone marrow aspirates from 54% of newly diagnosed MM patients with normal CC showed clonal anomalies by I-FISH. However, when BM samples contained 20% or more PCs, the percentage of detected anomalies by I-FISH rose to approximately 85%. A Mayo Clinic/Eastern Cooperative Oncology Group (ECOG) study using a FISH assay combined with an immunoglobulin (cIg) assay that stains PC cytoplasmic Ig light chains detected 60% IGH translocations compared with 11% detected by karyotyping, with all 11% showing t(11;14)(q13;q32) (Fig. 2).29 Based on those findings, the International Myeloma Working Group discourages the use of FISH on unselected BM cells and recommends the use of plasma cell–specific FISH.6 Using CC and PC FISH, laboratories can expect to find a PC clone in more than 90% of newly diagnosed MM patients.19,28
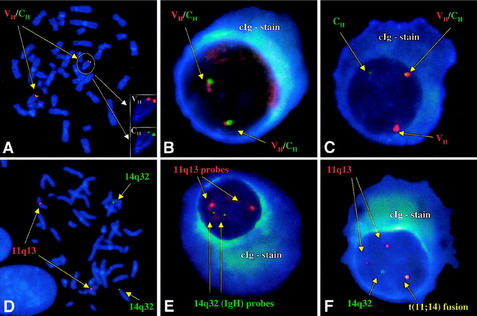
(Reprinted from Hayman SR, Bailey RJ, Jalal SM, et al. Translocations involving the immunoglobulin heavy-chain locus are possible early genetic events in patients with primary systemic amyloidosis. Blood 98:2266–8, 2001 [Fig. 1]; with permission.)
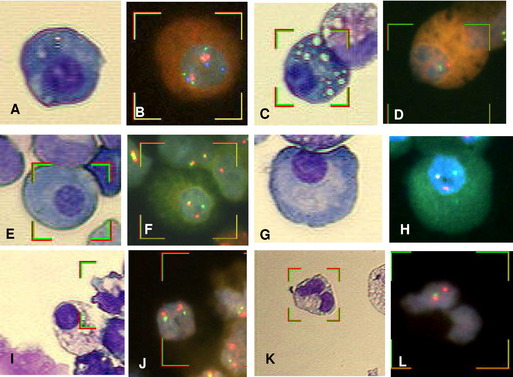
Identifying PCs for FISH analysis may be accomplished in any of three ways: (1) magnetic-activated cell sorting using anti-CD138 microbeads (purity approximately 90%) 19,30–35; (2) cIg-FISH36; or (3) morphology using sequential May–Grünwald Giemsa staining to identify malignant PCs by their characteristic morphology, followed by FISH (Fig. 3A–J).37 The last technique has the advantage of correlating cytogenetic aberrations with cell lineage. Correlating discrepant CC results with cell morphology allows for the detection of myeloid neoplasms coexisting with MM (see Fig. 3K,L).
The Mayo Clinic and the International Myeloma Working Group recommend the following FISH probes for risk stratification: –13/del(13q), del(17p)/TP53, and the two IGH high-risk translocations that are not clearly visible by CC, t(4;14)(p16;q32) and t(14;16)(q32;q23) (Fig. 4; see Table 2).15,18 Because hyperdiploidy and t(11;14)(q13;q32) are the most common aberrations observed in MM and define the standard risk group, many clinical laboratories test for them routinely.

Fig. 4 Genetic testing in newly diagnosed multiple myeloma for risk stratification.
(Adapted from Fig. 1 from Kumar SK, Mikhael JR, Buadi FK, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy [mSMART] consensus guidelines. Mayo Clin Proc 2009;84:1095–110; with permission.)
Other Molecular Assays
Information on copy number alterations in bone marrow samples can be detected with array comparative genomic hybridization (aCGH) or high-resolution single nucleotide polymorphism (SNP) arrays. These DNA-based arrays are highly effective in determining the chromosomal site, size, and genetic content of copy number alterations and defining the minimal region of gain or loss. Together with GEP, mutation analysis, and methylation studies, these genomic tools have the potential to identify candidate genes important in myeloma pathogenesis and to establish a new molecular risk stratification model. As with PC-FISH, these molecular assays yield the best results when purified PCs are used.18,30–35,38,39
Clinical Significance of Cytogenetic Aberrations
The first broad classification of MM is based on ploidy. Hyperdiploidy and non-hyperdiploidy represent the two major pathways for MGUS progression to MM (Table 3; see Fig. 1).7,11,24,29,40–45 The hyperdiploid subgroup comprises approximately 60% of MM patients and demonstrates better OS than the non-hyperdiploid subgroup. Key findings in the hyperdiploid subgroup include karyotypes with 48 to 74 chromosomes, frequent gains of odd-numbered chromosomes (especially 3, 5, 7, 9, 11, 15, 19, or 21), and compared with the non-hyperdiploid subgroup, less frequent structural aberrations and less frequent IGH translocations (∼10%–20%). Using high-resolution SNP arrays, the rank order of frequency of whole chromosomes gained in hyperdiploid MM is 15 > 9 > 5 > 19 > 3 > 11 > 7 > 21.33 In the non-hyperdiploid subtype, in contrast, the 40% of MM patients with generally poor OS share chromosome counts of 47 or less or 75 or greater and numerous structural aberrations, including frequent IGH translocations (∼70%).11,29,43–47 Chromosomes 13, 14, 16, and 22 are frequently lost in hypodiploid (defined as less than 45 chromosomes) MM. Near-tetraploid karyotypes are included in the non-hyperdiploid group because they appear to be doubling products of pseudodiploid or hypodiploid karyotypes.24,29,43,48
IGH Translocations Are Primary Aberrations in MM
IGH translocations targeting the IGH class switching region are reported in approximately 65% of patients with MM.29,42,49 Because IGH translocations are detected in MGUS and SMM, they are considered the primary “immortalizing” events, yet alone they are not sufficient for transformation.11 At the molecular level, these translocations juxtapose oncogenes into the proximity of the IGH enhancers, driving abnormal expression of the translocated oncogenes. Each translocation subgroup is associated with dysregulation of one of three cyclin D genes—CCND1, CCND2, or CCND3. CCND1 and CCND3 are directly overexpressed as the result of t(11;14) and t(6;14), respectively, while CCND2 is indirectly upregulated in the t(4;14) and MAF translocation group 7 (see Table 2). An additional approximately 20% of MM patients harbor IGH translocations involving other partner genes.29 The frequency of IGH translocations increases from MGUS to MM, suggesting some IGH translocations occur as secondary events in patients who present with hyperdiploid MM.29 In the absence of an IGH translocation, a diagnosis of κ and λ light chain immunoglobulin gene translocations, which have reported frequencies of up to 20% in MM, should be considered.11 The most common IGH translocation partners—4p16 (FGFR3 and MMSET), 11q13 (CCND1), 6p21 (CCND3), 16q23 (MAF), and 20q12 (MAFB)—are discussed in more detail in the text that follows, along with associated therapy and outcomes (see Table 3; Table 4). For a comprehensive discussion of the role of immunoglobulin gene rearrangements in antibody diversity and the pathogenesis of MM, see the review by Gonzalez and colleagues.9
Table 4 Correlation between MM therapy and cytogenetics
| Abnormality | Relationship with Treatment | References |
| Deletion 1p | Associated with short remission and survival in patients undergoing high-dose therapy and a single autologous transplant. | Qazilbash et al75 |
| Deletion of 1p21 or 17p | Adversely affects outcome for lenalidomide- plus dexamethasone-treated patients with relapsed or refractory MM. Co-presence of t(4;14) does not appear to be an adverse risk factor for PFS or OS. | Reece et al102 |
| Amplification of 1q | Thalidomide improved 5 year EFS in patient lacking amp(1q21) but not in patients with amp(1q21) and did not improve OS in either group. | Hanamura et al74 |
| del(17p13) or gain of 1q21 | Associated with a dismal OS when treated with lenalidomide and dexamethasone. | Klein et al104 |
| Absence of metaphase chromosome 13 aberrations and hypodiploidy | Favorable prognostic factor in tandem autologous SCTs. | Shaughnessy et al117 |
| t(4;14) and low TP53 expression | Bortezomib may overcome poor prognosis significance of t(4;14) but cannot overcome the adverse outcome of low TP53 expression or the high-risk 17-gene MM model. | Barlogie et al118 |
| Low TP53 gene expression and del(17p13) | Deletion is an independent adverse prognostic marker in newly diagnosed MM treated with autologous SCT. | Xiong et al32 |
| TP53 deletion | Patients with del17p/TP53 show an extremely poor outcome with VRD (lenalidomide, dexamethasone [RD] and bortezomib) combination. | Dimopoulos103 |
t(4;14)(p16;q32)
t(4;14)(p16;q32) occurs in approximately 15% of MM patients and results in the direct dysregulation of both the fibroblast growth factor receptor 3 gene (FGFR3) and a histone methyltransferase gene referred to as the MM SET domain gene (MMSET). MMSET is also known as WHSC1, or the Wolf-Hirschhorn syndrome candidate gene. Derivative 14 expresses FGFR3, an oncogenic tyrosine kinase receptor, and MMSET is overexpressed as a result of the der(4) fusion.45,49 Approximately 20% of t(4;14)-positive tumors lack FGFR3 expression, a finding that correlates with loss of der(14).50,51 The translocation results in the exchange of two subtelomeric light staining G-bands, so the best methods for detecting this IGH rearrangement are PC FISH and reverse transcription polymerase chain reaction (RT-PCR) for the hybrid IGH/MMSET transcript.29,52 The translocation has been reported in both asymptomatic and symptomatic disease.45,52 When observed in asymptomatic disease (MGUS and SMM), approximately 25% of cases progress to symptomatic MM in less than a year from diagnosis,53 and associated features include frequent IgA isotype (∼50%), concurrent 13q deletion (∼90%), high Sβ2M concentration (usually ≥2.3 mg/L), up-regulation of CCND2, and infrequent presentation with bone lesions.7,41,50,52–55 Although the overall initial therapy response rate for this subgroup is good, median progression-free survival is approximately 12 months, and more than one-third of patients experience rapid, aggressive, and drug-resistant relapses.53 High-dose chemotherapy with stem cell transplantation offers better rates of overall response and progression-free survival.53,55 In addition, emerging data using proteasome inhibitors (bortezomib) show promise for this poor-risk subgroup (see Table 4).53,56
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Syndromes
Syndromes
 Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


