Acute Myeloid Leukemia: Conventional Cytogenetics, FISH, and Moleculocentric Methodologies
Keywords
• AML • Cytogenetics • FISH • Molecular genetic • Classification
Acute myeloid leukemia (AML) is a heterogeneous group of malignancies that account for 1 of 4 broad subtypes of leukemia, neoplasms typically originating in the bone marrow and manifesting in the peripheral blood. AML is most common in the elderly population, with an incidence of approximately 3.4 per 100,000 per year,1 and a median age of 70 years at diagnosis. The morphologic hallmark of AML is an excessive accumulation (typically >20%) of blasts and other defined immature cells affecting 1 or more “myeloid” lineages, which includes myeloid, monocytic, erythroid, and megakaryocytic precursors. The fundamental defect leading to this accumulation is maturation arrest. AML historically has been categorized by not only the lineages that are affected but also by the stage and degree of differentiation at which the block occurred. The outcome of this block in the normal physiologic progression of maturation is loss of normal hematopoietic differentiation, with an accumulation of blasts, leading to thrombocytopenia, neutropenia, and anemia.
There have been different attempts to classify AML into reproducible, clinically relevant groups. The French American British (FAB) scheme classified the subtypes of AML based on morphologic, immunophenotypic, and cytochemical features, using designations from M0 to M7. This system was remarkably prescient, in that some subtypes were subsequently associated with specific recurrent chromosome abnormalities [eg, FAB M3 with t(15;17)]. More recently (initially in 2001, updated in 2008), the World Health Organization (WHO) has incorporated genetic features into the classification of subtypes of AML. Indeed, approximately 60% of AMLs are now genetically classified,2 although less than one-half of these are cytogenetically defined, highlighting the expansion of those characterized by molecular abnormalities that are not discernible by conventional cytogenetics. The WHO categorizes AML into 4 main groupings: (1) AML with recurrent genetic abnormalities, (2) AML with myelodysplasia-related changes, (3) therapy-related myeloid neoplasms, and (4) AML not otherwise specified.3 This review focuses on the first group and highlights the central role of cytogenetics in the second and third groups; throughout, the emphasis will be on initial diagnosis and not on the topic of tracking minimal residual disease.
In addition, other recurrent cytogenetic defects and many of the important submicroscopic molecular genetic defects will also be noted. Indeed, the development of AML requires multiple events with the suggestion of at least 2 hits necessary for leukemogenesis. The 2 pathways currently purported to be involved in the development of AML are those that are necessary for proliferation and survival (class I mutations) and others that prevent normal hematopoiesis by blocking differentiation (class II mutations).4,5 Examples of class I mutations include those affecting FLT3, KIT, and NRAS, whereas examples of class II mutations include t(8;21) and inv(16)/t(16;16).
The Role of Conventional Cytogenetic Studies and Fluorescence in Situ Hybridization
Cytogenetic abnormalities detected at clinical presentation are one of the main predictors of outcome in AML.2,6,7 Conventional cytogenetic (CC) studies have defined certain chromosomal rearrangements as favorable risk, standard risk, and high risk (Table 1).8–14 Allied to this, these studies play an essential role in contemporary AML diagnosis and classification. Although most of the disease-defining aberrations are detected by CC studies, fluorescence in situ hybridization (FISH) and sometimes also reverse-transcription polymerase chain reaction (RT-PCR) may also play a central role at diagnosis.
When FISH is performed at diagnosis, it is usually considered to be either an adjunct to conventional cytogenetic studies to unravel the components of complex translocations or a way to identify underlying abnormalities that are cytogenetically cryptic (Fig. 1). Thus, FISH is often useful at diagnosis to identify subtle abnormalities or to define breakpoints. Often, metaphase spreads from diagnostic marrows for AML can have poor morphology that can render certain rearrangements equivocal, such as some MLL translocations and deletions of 17p13. In such cases, FISH can be helpful in discerning questionable rearrangements that might alter classification and treatment options. For example, some cases of t(15;17) might be missed by CC studies, and only detected by FISH (or RT-PCR). Furthermore, there is also some evidence that FISH can have predictive value at diagnosis in AML. For example, TP53 deletions identified by FISH portend a poor response to chemotherapy.15
In general, interphase FISH correlates extremely well with abnormalities detected by CC. There is some evidence from a large prospective study in a series of older AML patients that some common rearrangements [inv(16), MLL rearrangements] in AML are missed by CC studies but readily identified by FISH.16 The Eastern Collaborative Oncology Group compared CC studies and FISH in 237 AML specimens and found that a probe panel to detect monosomy 5/deletion 5q, monosomy 7/deletion 7q, trisomy 8, t(8;21), t(9;22), MLL rearrangements with various partners, t(15;17), and inv(16)/t(16;16), had a concordance between 98% and 100%.17 In cases without sufficient metaphase cells for evaluation or metaphase cells with poor morphology or those with nonevaluable chromosomes, FISH studies can be extremely helpful in defining specific entities and the determination of cytogenetic risk groups.
Spectral Karyotyping, Multicolor FISH, and Combined Binary Ratio FISH
Spectral karyotyping (SKY), multicolor FISH (M-FISH), and combined binary ratio FISH (COBRA FISH) are molecular cytogenetic techniques that permit simultaneous visualization of all chromosomes in a different color by combinatorial chromosome painting, using slightly different methodologies.18 This group of techniques is useful in the detection of translocations, unbalanced rearrangements, and complex karyotypes, in which the resolution of G-banding may be too poor to detect all the rearrangements present. Because these are performed on a background of metaphase chromosomes, they allow the visualization of chromosomal rearrangements, including changes in copy number and location. However, none of these is currently used in most diagnostic laboratories.
Multiplex PCR
The combination of cytogenetic and molecular methods for detection of translocations in AML has multiple benefits and should be considered at diagnosis to detect common rearrangements. Numerous platforms are available that target the common recurrent cytogenetic abnormalities seen in AML, with the benefit of detection of cryptic rearrangements.19 The technique of multiplex RT-PCR for translocation detection has the advantage of shorter turnaround time and no requirement for dividing cells in the diagnosis of AML, both of which are essential for conventional cytogenetics.
AML with Recurrent Genetic Abnormalities
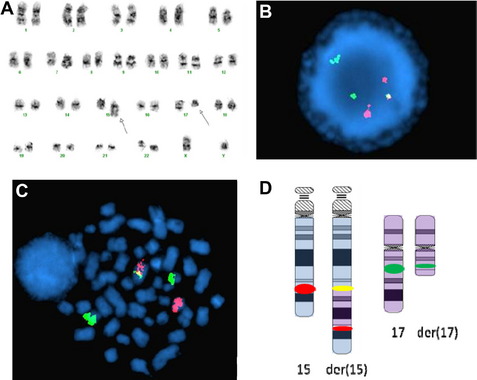
There are currently 7 chromosome rearrangements that fall under the category of AML with recurrent genetic abnormalities. They are typically balanced translocations or inversions and are identified in 20% to 30% of all cases of AML. Often these aberrations seemingly occur as the sole cytogenetic abnormality, but they can be seen in the background of complex abnormalities or molecularly detected mutations, which, in some cases, alter risk stratification. In the 2001 edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, four such translocations were formally integrated into the diagnosis of AML, and in 2008 edition they were modified and increased to seven (Table 2). These balanced rearrangements usually lead to fusion genes and chimeric protein expression and are considered necessary but not sufficient for development of AML.20
t(8;21)(q22;q22.3)
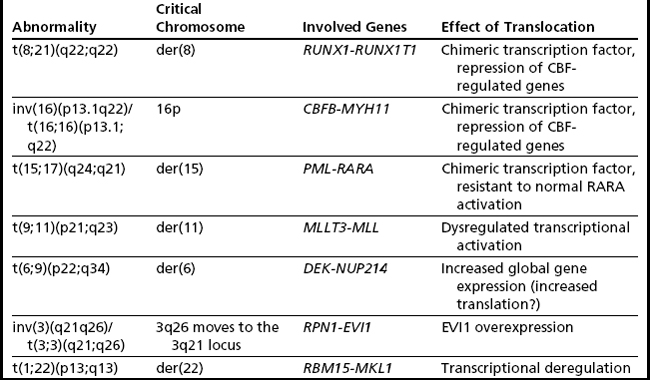
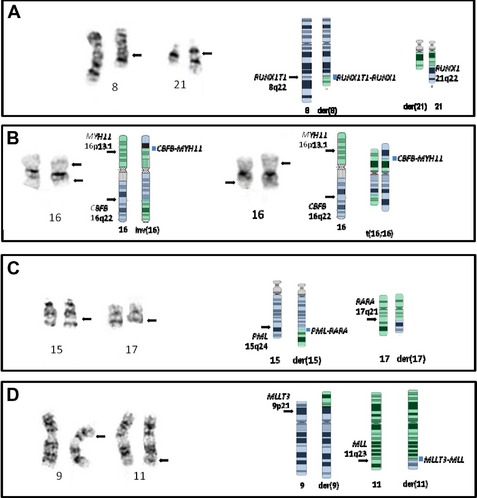
The t(8;21) involves RUNX1T1 on 8q22 and RUNX1 on 21q22 (Fig. 2A) (formerly ETO and AML1/CBFA2, respectively), and this type of AML is associated with a favorable prognosis in children and adults.2,21 There is some correlation with the previously designated FAB type of M2 (myeloblastic with maturation), especially those with characteristic blastic morphology (delicate Auer rods, salmon-pink granules) and coexpression of a number of B-cell antigens. The t(8;21) is seen in approximately 5% to 7% of de novo AML in adults. Of note, cases with a t(8;21)(q22;q22) are considered to be AML regardless of the percentage of blasts in the bone marrow (ie, it is acceptable to diagnose AML with <20% blasts). Although this is an easy translocation to identify cytogenetically, recognition can be complicated by a complex karyotype or a cryptic insertion. In approximately 3% to 4% of t(8;21)-positive cases, the translocation is complex, involving 8q22, 21q22, and another chromosome22,23 with evidence indicating that the derivative chromosome 8 is the chromosome harboring the critical abnormal fusion. Secondary abnormalities include sex chromosome loss (X in females, Y in males) and deletions of the long arm of chromosome 9. There appears to be no significant difference in overall survival in patients with a deletion of 9q or loss of an X chromosome; however, loss of the Y chromosome is associated with better overall survival.2 Individuals with t(8;21) AML have a complete remission rate of 97% and a 10-year overall survival rate of 61%.
RUNX1 encodes the core binding factor subunit alpha 2, with the fusion gene leading to the transcriptional repression of genes that are normally physiologically activated by RUNX1, preventing granulocytic differentiation through dominant-negative inhibition.24 Although the t(8;21) appears to be necessary for AML development, it is not sufficient, in that additional mutations are necessary for development of this form of AML. This is not unique to t(8;21) AML and is operative in other AMLs; however, this phenomenon is perhaps best studied in this context. This has been demonstrated in a number of ways, including the identification of the t(8;21) in utero followed by a latency of up to 10 years before the development of AML.25 There is also evidence that the RUNX1T1-RUNX1 transcript persists in patients in long-term, sometimes decades-long, remission,26,27 although these patients are more likely to relapse than those in whom the transcript is undetectable molecularly.28 Interestingly, even after relapse, t(8;21)-positive leukemias maintain their good prognosis and can respond well to bone marrow transplantation.
Several factors alter the favorable prognosis of t(8;21) AML. Recently, identification of a more potent form of RUNX1T1-RUNX1 was identified, which shows enhanced leukemogenic potential caused by alternative splicing of exon 9a of the fusion transcript.28 Although the exon 9 splice variant was seen in patients with RUNX1-RUNX1T1 rearrangements, the patients who remained in complete remission were found to have a faster disappearance of the RUNX1-RUNX1T1 exon 9a splice variant compared with patients bound to relapse. Mutations in the KIT gene, a receptor tyrosine kinase, have been identified in a subset of patients with t(8;21). Several studies have associated the presence of KIT-activating mutations (most commonly affecting an aspartate residue at codon 816) with poorer prognosis in adult patients with this translocation.29,30 However, these mutations appear not to be prognostically relevant in pediatric patients.31
inv(16)(p13.1q22) or t(16;16)(p13.1;q22)
These molecularly identical events involve CBFB on 16q22 and MYH11 on 16p13.1 (formerly PEBP2B and SMMHC/SMHC, respectively), and this form of AML is usually, but not invariably, associated with acute myelomonocytic leukemia with abnormal eosinophils harboring basophilic granules (previously designated FAB M4Eo). Such rearrangements of chromosome 16 are seen in approximately 5% to 8% of all cases of adult AML, with the inversion much more common (95%) (see Fig. 2B, left) than the translocation (5%) (see Fig. 2B, right).3 As with the t(8;21)(q22;q22) above, it is possible to diagnose AML when this translocation/inversion is present, regardless of the percentage of blasts.
The inv(16)/t(16;16) is the sole abnormality in 70% of cases. The most frequent secondary changes, trisomy 22, trisomy 8, deletion 7q, and trisomy 21 do not alter the favorable prognosis of this rearrangement; indeed, there is evidence that +22 improves overall outcome.2 Approximately 20% of cases harbor KIT mutations in exons 8 and 17, and this is associated with a less-favorable outcome.32,33 RAS mutations occur in approximately 36% of cases, with NRAS more commonly mutated than KRAS (32% vs 6%); however, these are not currently considered prognostically significant.34 Individuals harboring inv(16)/t(16;16) have a complete remission rate of 92% and a 10-year overall survival rate of 55%.2
Physiologically, the RUNX1 protein, genetically disrupted in the t(8;21), and CBFB, genetically disrupted here, associate to form core binding factor (CBF), which is a key transcription factor central to the regulation of normal hematopoiesis, via its ability to positively control the expression of a variety of hematopoietically important genes.20,35 Like RUNX1-RUNX1T1, the CBFB-MYH11 fusion gene product acts in a dominant-negative fashion by preventing normal heterodimer formation36 leading to a differentiation block. Thus, CBFB-MYH11 and RUNX1T1-RUNX1 AMLs can be considered together as CBF-AMLs; interestingly, they have similarly good outcomes.
t(15;17)(q24;q21)
The t(15;17) is essentially synonymous with the diagnosis of acute promyelocytic leukemia (APL), which is equivalent to the entity designated by FAB as AML M3 (see Fig. 2C). The translocation was one of the first balanced translocations described in acute leukemia.37 The breakpoint on chromosome 15 has recently been modified from 15q22 to 15q24.1 (UCSC Human Genome Browser, February 2009, hg19).38
It is important to recognize this translocation quite rapidly because of the need to begin (targeted and specific) treatment due to the high rate of the development of disseminated intravascular coagulation in APL. t(15;17) AML comprises about 5% to 13% of all cases of AML and is seen as the sole abnormality in approximately 75% of cases. As above for the 2 CBF AMLs, it is possible to diagnose AML (specifically APL) if this translocation is present, even if there are less than 20% blasts/leukemic promyelocytes. The most common additional abnormalities include trisomy 8, deletion 7q, deletion 9q, and an isochromosome of the derivative 17q from the t(15;17). However, these additional chromosome rearrangements, including those that would typically be considered adverse (eg, abnormalities of 17p and complex abnormalities), do not alter the excellent prognosis of APL.2 Individuals with a t(15;17) AML have a 93% complete remission rate and a 10-year overall survival rate of 81%.2
FLT3 mutations in APL are seen in 40% of cases and are associated with higher white blood cell count and the morphologic hypogranular variant. However, these mutations have not been shown to have a significant impact on treatment sensitivity, remission status, or overall outcome in APL.39
t(9;11)(p22;q23)
This translocation involves MMLT3 on 9p22 and MLL on 11q23 and is associated with AML with monocytic features (see Fig. 2D). The t(9;11) constitutes one-third of all MLL rearrangements in AML.40 It is more common in pediatric AML (10%)41 than in adult AML (2%). Trisomy 8 is the most common secondary abnormality (20% of cases), with trisomy 6, 19, and 20 also seen with t(9;11), but none appear to affect the association of this translocation with an intermediate prognosis. This form of AML is considered to have a better prognosis than AML with other translocations involving MLL (and for this reason it was separated out in WHO 2008) and has an overall 10-year survival rate of approximately 39%.2
inv(3)(q21q26)/t(3;3)(q21;q26)
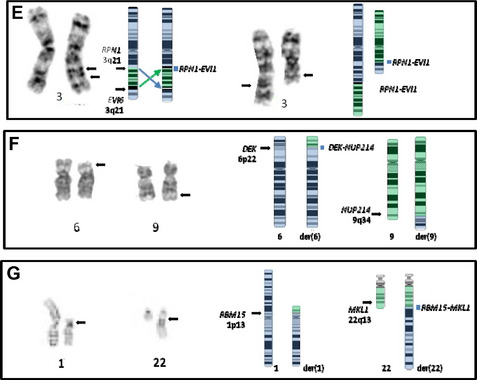
These 3q rearrangements are seen in 1% to 2% of cases of AML. Unlike the aforementioned translocations that result in fusion genes and chimeric proteins, the consequence of this genetic event is dysregulated (over)expression of EVI1. Multi-lineage dysplasia and megakaryocytic differentiation are common features, and the prognosis is typically poor. The inversion (see Fig. 2E, left) is at least twice as common as the translocation (see Fig. 2E, right), and rare cryptic rearrangements have been identified. The most common secondary abnormality is monosomy 7 (approximately 50% of cases), and this worsens the already dismal poor prognosis associated with this rearrangement.42 This is not unexpected, because this combination meets criteria for a monosomal karyotype (see later discussion). Deletions of 5q, trisomy 8, and complex karyotypes can also be seen with these 3q rearrangements. However, these additional abnormalities do not influence the extremely poor outcome of these patients who have a 10-year overall survival rate of 3%.2
t(6;9)(p22;q34)
This translocation (see Fig. 2F) is seen in 1% to 2% of cases of AML, which often have a basophilic component. These cases have a poor prognosis; this is particularly dismal in pediatric patients, with some reports of 0% overall survival rate at 4 years,43 whereas the 10-year overall survival rate in adults is 27%.2 The genes involved are DEK at 6p22 and NUP214 (CAN) at 9q34 and are the sole abnormality in approximately 80% of cases.44 When additional abnormalities are seen, trisomy 8, del(12p), and trisomy 13 are the most common; however, their effect on outcome is unclear. More than 70% of cases with a t(6;9) also contain the FLT3 internal tandem duplication, with the combination associated with a high white blood cell count and an even poorer prognosis.44,45
t(1;22)(p13;q13)
This translocation fuses RBM15 (formerly OTT) at 1p13 with MKL (formerly MAL) at 22q13 (see Fig. 2G)46 and is seen in less than 1% of AML.2 The translocation is most often seen in infants or young children, with most cases being observed in the first 6 months of life. The t(1;22) is associated with acute megakaryoblastic leukemia (AMKL) and is the most common abnormality seen in pediatric AMKL in the first year of life. Although the prognosis of AMKL is poor, infants with the t(1;22) fare better than those lacking the translocation.3 The t(1;22) has been associated with an intermediate outcome, with a 3-year event-free survival rate of approximately 50%. This is the sole abnormality in 60% of cases (mostly those under 6 months of age), with older children more likely to have a hyperdiploid karyotype [2, +19, +der(1)t(1;22), +6, +21]. These additional changes are reported to have no effect on outcome. The presence of supernumerary der(22)t(1;22) and the retention of the der(22) as the sole abnormality in some cases suggest that the critical event lies on the der(22), leading to the fusion gene RBM15-MKL.
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Myeloma: Current Perspectives
Myeloma: Current Perspectives
 Syndromes
Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


