The myelodysplastic syndromes (MDS) comprise a spectrum of neoplastic hematopoietic stem cell disorders that share the features of ineffective and dysplastic hematopoiesis accompanied by peripheral blood cytopenias. Subclassifications of MDS can be made based on differences in hematopoietic lineage affected (e.g., erythroid, myeloid, and megakaryocytic), percentage of bone marrow myeloblasts, and molecular abnormalities. The incidence of MDS in the United States is estimated at 3.3 per 100,000.
1,2 MDS most often affects older individuals. In the population >60 years of age, the incidence of MDS is estimated at 50 per 100,000. Predisposing causes of MDS include prior exposure to organic solvents such as benzenes, ionizing irradiation, chemotherapy, and tobacco smoking.
3 Most cases of MDS occur de novo with a rising fraction secondary to prior therapy such as chemotherapy (particularly alkylating agents) and radiation therapy. The natural history of MDS is highly variable and likely related to the myriad of genetic, epigenetic, and microenvironmental changes that characterize each individual’s disease. In patients who develop therapy-related MDS, the disease is often characterized by adverse cytogenetic abnormalities, an aggressive clinical course, and rapid transformation into acute myeloid leukemia (AML). Approximately 30% of MDS cases progress to AML, and for that reason, this syndrome has been historically regarded as a
preleukemia. However, most patients with MDS succumb to infection and other complications related to cytopenias.
4
PATHOGENESIS
MDS arises from genetic or epigenetic changes in hematopoietic stem cells or as a result of aberrant growth of a stem cell under adverse microenvironmental pressures.
5,6,7 Subsequent DNA damage events and immunologic responses that promote survival of the dysplastic stem/progenitor cell and clonal dominance may occur.
6,8 Genomic instability, because of an accumulation of genomic damage and failure to repair these sites of damage, is a hallmark feature of MDS pathology. Another factor contributing to genomic instability is age-related telomere erosion arising from senescence-related DNA replication stress and impairment in telomere repair.
9 Early in the disease course, the MDS bone marrow reveals impaired differentiation and increased apoptosis.
10 As the MDS progresses, the bone marrow shows increased cell proliferation and accumulation of immature stem/progenitor cells. Eventually, a clone of mutated stem/progenitor cells loses responsiveness to apoptosis signaling, resulting in accumulation of neoplastic myeloblasts associated with a diagnosis of leukemia.
In contrast to leukemias like chronic myeloid leukemia and acute promyelocytic leukemia (APL), where a distinguishing genetic mutation accounts for pathogenesis of the disease, MDS can present with variable and heterogeneous genetic or epigenetic changes. This variability is likely the reason for the syndrome’s heterogeneity in clinical presentation and bone marrow pathology. In MDS, certain chromosomal abnormalities present more frequently. Among the most common include structural or numerical deletions of chromosomes 5, 7, and 20, and in men, the deletion of Y chromosome. Identifying cytogenetic abnormalities in MDS is a key determinant of diagnosis and prognosis, providing genetic confirmation of diagnosis, predicting tempo of disease progression, and assisting in the selection of appropriate therapeutics. Unfortunately, because of technical limitations of commercially available metaphase cytogenetics (i.e., Giemsa band staining of whole chromosomes), approximately 30% to 60% of MDS patients have normal karyotype. This group of normal cytogenetic MDS patients is clinically heterogeneous in presentation, disease behavior, and response to therapy, which likely reflects the heterogeneous changes at the molecular level.
With the majority of MDS patients showing no cytogenetic alteration in their MDS clone, candidate gene approaches have been employed to search for recurring gene mutations. Investigations of select genes (e.g.,
RUNX1,
NPM-1,
FLT3,
TP53, and rat sarcoma (Ras) pathway) have found gene mutations in MDS patients with normal karyotype; however, these select mutations were present in only a minority (<5%) of patients. By studying the whole genome of MDS cells using single-nucleotide polymorphism (SNP) array and genome sequencing, investigators found acquired deletions, and missense and nonsense mutations in
TET2 in 26% of MDS patients, representing the most frequently reported gene abnormality in MDS.
11 In cases where a TET2 mutation was found the abnormality was found in nearly all bone marrow cells including CD34+ stem/progenitor cells. These loss of function abnormalities of
TET2 are believed to be a first hit in the pathogenesis of MDS. Functionally, TET2 is involved in regulating DNA methylation.
12 Perturbations in TET2 result in dysregulated methylation (both hypomethylation and hypermethylation). Thus, it has been proposed that treatment of
TET2 mutant MDS with hypomethylating agents may bring about improved outcomes. An early study corroborates this hypothesis by showing higher complete remission (CR) rates after azacitidine in MDS patients with
TET2 mutation compared with those with wild type
TET2 (65% vs. 30%,
P = .01),
13 although, there was no difference in survival detected.
The 5q-syndrome is a discrete clinical and pathologic subset of MDS characterized by an isolated chromosome 5q deletion, a hypoplastic macrocytic anemia, normal to elevated platelet count and pathologically characterized by the presence of hypolobulated megakaryocytes. In del(5q) MDS, there is a commonly deleted region (CDR) on chromosome 5; a 1.5 Mb interstitial segment that extends between 5q31 and 5q32 that contains 44 candidate genes.
14 Using a functional RNA interference screen for each of the genes in the CDR, only knock down of the
RPS14 gene yielded an erythroid phenotype similar to the 5q-syndrome. RPS14 is a ribosomal protein which is a component of the structural framework of the 40s ribosomal subunit. Interestingly, mutational inactivation of
RPS19, another gene involved in ribosomal biogenesis, is implicated in the pathogenesis of Diamond-Blackfan anemia. Evidence from animal models shows that impaired ribosome biogenesis leads to p53 stabilization and activation that contributes to the hypoplastic anemia and increased apoptosis of erythroid precursors in del(5q) MDS.
Epigenetic changes in MDS, such as aberrancies in DNA methylation and/or histone acetylation, are also believed to contribute to the pathogenesis of MDS. Increased methylation of CpG dinucleotides concentrated in the promoter regions of some genes (also known as CpG islands) leads to silencing of the gene. In MDS, increased methylation of CpG islands has been reported with more advanced MDS displaying greater methylation than lower risk disease. DNA transcription can also be affected by how condensed the DNA is wrapped around histone cores. When histone cores are acetylated, the negatively charged acetyl groups neutralize the positive charge on the histone proteins, resulting in the release of the negatively charged DNA. As a consequence, the condensed chromatin relaxes and permits higher activity of gene transcription. A balance exists between histone acetyltransferases (HAT), which open gene transcription, and histone deacetylases (HDAC), which close gene transcription. Abnormalities in HAT and HDAC have been reported in patients with MDS.
15,16The MDS microenvironment also contributes to the pathobiology of disease. The abnormal bone marrow microenvironment is believed to promote the survival and proliferation of neoplastic MDS cells. Perturbations of several cytokines in bone marrow and peripheral blood have been reported, including IL-6, IL-8, stem cell factor, erythropoietin, TGF-/β, and TNF-α<x
17,18,19,20,21 Interestingly, apoptosis is a prominent finding in early MDS compared with late MDS.
10,22 Moreover, apoptosis occurs in nonclonal cells, suggesting a collateral damage that impairs the bone marrow’s ability to support effective hematopoiesis.
23Another unique aspect of the supportive MDS bone marrow microenvironment is heightened neovascularization. Patients with MDS exhibit increased blood vessel density in their bone marrow.
24,25,26,27 Furthermore, abnormal elevations in angiogenic cytokines such as VEGF, bFGF, and angiopoietins have been reported in MDS bone marrow.
25,28 Given that myelomonocytic precursors from MDS patients overexpress VEGF and VEGFR2, paracrine and autocrine loops are believed to further stimulate MDS cell proliferation while suppressing effective erythropoiesis.
There is mounting evidence that immune dysregulation plays an important role in MDS pathophysiology. First, the incidence of autoimmune disorders is increased in patients with MDS.
29 Additionally, MDS can share disease characteristics that overlap with aplastic anemia and large granular lymphocytic leukemia, which are known to involve autoreactive T lymphocytes. Clinically, immunosuppression with antithymocyte globulin, cyclosporine and prednisone are effective in select groups of patients with MDS. Important questions relating to the mechanisms of autoimmunity in MDS remain to be answered.
PROGNOSIS
A major shortcoming of past and present classification systems for MDS is the wide variation in patient outcomes within diagnostic categories. For example, although it is understood that patients with refractory anemia with excess blasts (RAEB) have worse outcomes than patients with refractory anemia and ringed sideroblasts (RARS), there is still considerable heterogeneity of clinical outcomes among RARS patients. To accurately forecast outcomes for all MDS patients, prognostic scoring systems were developed. In 1997, evolution of prognostic tools and advancement of cytogenetic technology led to the landmark International Prognostic Scoring System (IPSS), which is the most widely used prognostic system for MDS patients (
Table 45-2).
31 This scoring system considers the number of peripheral blood cytopenias (with more lineages affected
indicating more advanced disease), enumeration of myeloblasts in the bone marrow (with more indicating advanced disease), and assessment of cytogenetic abnormalities. Based on these three disease characteristics at the time of diagnosis, an overall IPSS score can be assigned to the patient’s disease (
Table 45-2). The IPSS score is important for determining the patient’s risk for reduced survival and progression to AML, with higher risk groups at increased risk of disease progression and death. The IPSS is a useful tool for determining who and when to administer disease-altering therapy.
Despite the utility of the IPSS, it has several important limitations. First, the IPSS was only validated in previously untreated patients with de novo MDS and nonproliferative CMML. In addition, the IPSS, which was created in 1997, included MDS patients with blasts >20% (currently assessed as AML by WHO 2008 criteria). Thus, the high-risk IPSS group may have worse outcomes by including patients with more advanced disease. Given evidence that need for transfusion is a strong predictor of poor outcome in MDS patients,
32 another weakness of the IPSS is that it does not distinguish the severity of cytopenia (e.g., cytopenias so low that the patient requires transfusions vs. minimally decreased blood count with no need for transfusions). Finally, with further advancements in molecular genetic technology and better appreciation for MDS pathobiology, the limited number of cytogenetic abnormalities listed in the original IPSS (just five) are not sufficiently inclusive.
As a consequence, several next generation prognostic tools have been proposed and their validity are being tested. These newer prognostic systems build upon the WHO classification system and the 1997 IPSS by including additional disease characteristics such as need for transfusions, fibrosis in the bone marrow (
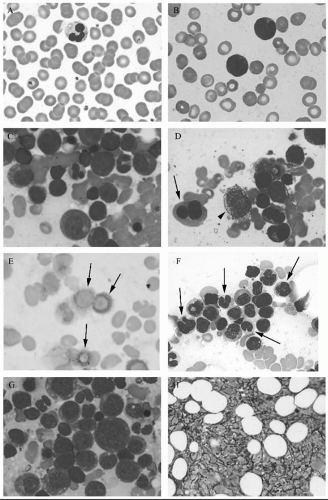
Fig. 45.1H), CD34+ cell clustering, ferritin level,
β-2- microglobulin level and new, recurrent DNA mutations.
33,34,35 For example, by combining WHO classification, IPSS score, and need for transfusions, the WHO-based prognostic scoring system was developed.
33 Moreover, this new tool has been validated for use throughout the MDS patient’s lifetime, allowing adjustment in disease expectation and not just limited to the time of diagnosis like the IPSS. Another new prognostic tool for MDS patients focuses on the IPSS lower-risk group (low/intermediate-1), which contains a heterogeneous group of individuals with different severities of cytopenias.
34 This new scoring system takes into account age, cytogenetic abnormalities, anemia, severity of thrombocytopenia and myeloblasts in the bone marrow, and stratifies lower-risk patients into those with favorable versus unfavorable prognosis. Use of this prognostic tool identifies lower-risk MDS patients who may benefit from therapies that bring about hematologic improvement (HI) alone or extend survival.
MANAGEMENT
At clinical presentation, precise disease classifications and prognostic tools are available to evaluate MDS patients. However, each individual brings his or her own unique condition. For example, sometimes bone marrow pathology may not fit cleanly in a WHO classification. Sometimes a crossover diagnosis between MDS and another disease, such as aplastic anemia or myelofibrosis, seems more appropriate than strictly limiting the diagnosis to one type. In recognition of the possibility of overlap
syndromes, the WHO classification systems have included the overlap subgroup, myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN). This subgroup includes CMML, atypical chronic myeloid leukemia, juvenile myelomonocytic leukemia and introduced in 2008, the provisional entity, RARS with thrombocytosis. These myeloproliferative and dysplastic bone marrow conditions seem to have in common aberrancies in the as-Mitogen-Activated Protein Kinas (MAPK) signaling pathway.
Occasionally, a confounding disease like an autoimmune disease (e.g., systemic lupus erythematosis) or chronic infection (e.g., HIV) may present with cytopenias and a bone marrow consistent with WHO MDS classification. In this situation, the clinician must balance the probabilities of the correct diagnosis. More commonly, the MDS patient’s performance status and comorbid diseases place boundaries on the intensity of treatment. This is especially true in MDS where the median age at presentation is 76. Finally, interpreting treatment results from the published medical literature and applying it to the presenting MDS patient can be difficult. Nuances in clinical trial design necessitate that clinicians understand each trial’s patient selection, assignment of therapy, and criteria for response.
Principles of Treatment Selection
Because of the heterogeneity of MDS, choice of initial therapy is based on several factors, such as chromosomal or molecular abnormalities, disease risk category, patient comorbidities, and performance status. Patient age may also be considered but is often a surrogate for comorbidities and organ reserve.
Consensus Guidelines
Three expert working groups have convened to define MDS treatment guidelines for practicing clinicians: the National Cancer Center Network (NCCN) in the United States, the British Committee for Standards in Haematology (BCSH) in the United Kingdom, and the Italian Society of Hematology (SIE) in Italy. Each working group employs different methods to derive their recommendations.
The NCCN/US guidelines are developed by consensus of expert opinion and quality of data, base treatment decisions on IPSS risk category, and present choices as flow charts.
36 The BCSH/UK guidelines are based on consensus and systematic literature review, base treatment on disease risk category (IPSS or Sanz), and also present choices as flow charts.
37 The SIE/Italian guidelines are constructed from consensus of experts and systematic literature review, and are presented as graded responses to relevant questions in MDS management.
38In general, the three working groups present similar recommendations for the diagnosis and management of MDS. In particular, the proposed initial evaluation and classification schemas are similar. For patients with lower-risk MDS, supportive care and growth factor support are recommended. For patients with intermediate-2 and high IPSS risk MDS, allogeneic hematopoietic cell transplantation (if donor available) and azanucleoside therapies are recommended. For patients with low and intermediate-1 IPSS risk MDS with a sole deletion of chromosome 5q, lenalidomide is recommended. The groups also recommend consideration of immunosuppressive therapy for patients <60 years, those with hypoplastic MDS, MDS with PNH, and MDS with HLA-DR15 phenotype. Also, common among the groups is the recommendation to consider iron chelation in low and intermediate-1 IPSS risk MDS patients.
Despite the mostly consistent recommendations among the groups, there are some differences. The BCSH/UK guidelines have not been updated since 2003, thus lack recommendations based on recent studies such as azanucleoside and immunomodulatory agents. Whereas the NCCN/US makes a preferred recommendation for 5-azacitidine (AZA, Vidaza) over 2′-deoxy-5-azacitidine (decitabine, DAC, Dacogen) based on survival data for AZA in patients with higher-risk disease,
39 the SIE/Italian group only recommends AZA and makes no recommendation for DAC. The groups also differ in first choice of iron chelating agent: the NCCN/US panel recommends subcutaneous deferoxamine (DFO), whereas the SIE/Italian group recommends oral deferasirox (Exjade) as the first-choice drug. In addition, the SIE/Italian group recommends using ferritin levels not as a decisive parameter to start chelation: rather as a marker of efficacy in the follow-up of chelation therapy.
Notably, where the groups cannot make a recommendation, largely because of lack of data, is whether a remission should be achieved in patients who intend to receive a transplant.
Response Criteria
Incorporating new therapeutic agents into the management of MDS should be based on convincing evidence of the intervention’s efficacy balanced by its safety. Moreover, when comparing therapeutic agents, similar response criteria are required for valid examination. Thus, precision in describing disease response is of crucial importance. Toward this end, an International Working Group (IWG) has created standardized
response criteria for MDS published originally in 2000 and revised in 2006.
40,41 These criteria include responses relative to altering natural history of disease, cytogenetic responses, HIs, and quality of life measures (
Table 45-3).