Myelodysplastic Syndromes
Olga Pozdnyakova, MD, PhD
Robert P. Hasserjian, MD
The myelodysplastic syndromes (MDSs) are a heterogeneous group of disorders caused by a clonal expansion of hematopoietic stem cells in which maturation is abnormal1 and cell production is ineffective, resulting in various degrees of cytopenia with anemia being the most common.2 MDS usually occurs in older adults with the median age at diagnosis of 65 years.3 Although the risk of developing MDS increases with age,4 therapy-related MDS can develop at any age and MDS can rarely affect children.5 MDS has been associated with exposure to environmental factors (including chemotherapy and radiation), certain inherited genetic abnormalities, and preceding acquired hematologic conditions (Table 7.1).
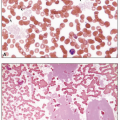
Many patients are asymptomatic and present with cytopenia(s) on routine blood count analysis. Some patients may present with symptoms and/or complications from a previously unrecognized cytopenia, such as infection, bleeding, easy bruising, and general fatigue.2 Complete blood count analysis typically shows normocytic or macrocytic anemia that is usually associated with inadequately low reticulocyte production and increased red blood cell distribution width reflecting anisocytosis. Anemia can be isolated or associated with leukopenia and/or thrombocytopenia; MDS only rarely presents with leukopenia and/or thrombocytopenia in the absence of anemia.6 The bone marrow is typically hypercellular for age despite peripheral blood cytopenias.7 In addition to the quantitative abnormalities described above, the cells in peripheral blood and bone marrow demonstrate dysplasia that involves one or more hematopoietic lineages (Table 7.2). Erythrocytes may show macrocytosis, anisocytosis, basophilic stippling, and Pappenheimer bodies.8 Poikilocytosis is usual and includes target cells, acanthocytes, elliptocytes, stomatocytes, red cell fragments, and teardrop cells (Fig. 7.1). Erythroid precursors in the bone marrow may show nuclear and/or cytoplasmic abnormalities. The former consist of budding, internuclear bridging, the presence of more than one nucleus per cell, karyorrhexis (fragmentation), abnormal chromatin (either fine or dense), and megaloblastic changes, in which the nucleus is enlarged and less mature than would be expected based on the degree of cytoplasmic hemoglobinization (Fig. 7.2).9 Cytoplasmic abnormalities include vacuolization and ring sideroblasts, which are erythroblasts that, on iron stain, contain at least five ferritin granules encircling at least one-third of the nucleus (Figs. 7.3 and 7.4).
Evidence of abnormal granulopoiesis on the peripheral smear includes nuclear hypersegmentation or hyposegmentation, Döhle bodies, decreased or absent cytoplasmic granules, and the presence of immature cells, including blasts (which should not exceed 20% of the leukocytes) (Fig. 7.5).1,10 The nuclei may exhibit dense chromatin clumping, and many are hypolobulated, with a single lobe or two joined by a thin band of chromatin, resembling the congenital Pelger-Huët anomaly (Fig. 7.6).11 Such pseudo Pelger-Huët cells, if frequent on a blood smear, strongly suggest an underlying MDS. Eosinophils and basophils may have diminished granules and/or decreased nuclear lobulation (Fig. 7.1). In the bone marrow biopsy, granulopoiesis may be architecturally disturbed whereby blasts and immature myeloid precursors occur centrally in the marrow space rather than in their typical paratrabecular location, a finding designated as abnormal localization of immature precursors (ALIP) (Fig. 7.7).12
Abnormal thrombopoiesis on the peripheral smear is manifested by giant platelets and forms with decreased or absent granules. In the bone marrow, dysplastic megakaryocytes are small (micromegakaryocytes), and possess abnormal nuclei that are multiple and widely separated or have decreased or absent lobulation (Figs. 7.8 and 7.9).11,13 In all hematopoietic lineages, the dysplasia should affect at least 10% of the cells within the lineage to be considered significant according to the WHO Classification criteria.14,15
Morphologic dysplasia can be complemented by evaluation of “immunophenotypic dysplasia” by multiparameter flow cytometric (FCM) analysis, especially in patients with borderline/minimal dysplasia and low blast counts. The International/European LeukemiaNet Working Group for Flow Cytometry in MDS has developed an FCM MDS score
based on a simple approach using the following widely applicable parameters/antigens: side scatter cell properties, CD10, CD34, and CD45 (Table 7.3; Fig. 7.10). A FCM score of at least 2 has high specificity for an MDS diagnosis and may be adopted as a basic protocol for the diagnostic workup of MDS patients.16,17 These parameters may be supplemented by the analysis of altered expression of additional antigens on myeloblasts, maturing myeloid cells, and monocytes (Table 7.4; Fig. 7.11). However, until uniform FCM diagnostic criteria for MDS are developed, reference ranges for each parameter should be validated based on individual laboratory practices.
based on a simple approach using the following widely applicable parameters/antigens: side scatter cell properties, CD10, CD34, and CD45 (Table 7.3; Fig. 7.10). A FCM score of at least 2 has high specificity for an MDS diagnosis and may be adopted as a basic protocol for the diagnostic workup of MDS patients.16,17 These parameters may be supplemented by the analysis of altered expression of additional antigens on myeloblasts, maturing myeloid cells, and monocytes (Table 7.4; Fig. 7.11). However, until uniform FCM diagnostic criteria for MDS are developed, reference ranges for each parameter should be validated based on individual laboratory practices.
Patients with MDS should always undergo genetic evaluation that not only helps establish disease clonality and classify MDS subtypes but also determines prognostic risk group and helps guide therapy.18 The presence of certain recurrent cytogenetic abnormalities, as detected by routine karyotype, in patients with cytopenia is considered MDS-defining even in the absence of significant dysplasia (Table 7.5).14,15 Recent technological advances in high-throughput genomic sequencing have revealed a panoply of MDS-specific somatic mutations.19 Up to 90% of MDS patients demonstrate at least one acquired somatic mutation compared to a much lower (50% to 60%) rate of abnormalities detected by conventional karyotype. Mutations in more than 40 genes have been identified in MDS that fall into several general categories, including transcription factors, epigenetic regulators and chromatin-remodeling factors, pre-mRNA splicing factors, and signaling molecules (particularly in the RAS pathway) (Table 7.6).20,21 Detection of MDS-specific somatic mutations has drastically changed the current understanding of MDS biology and has the potential to improve diagnostic criteria, prognostication, and monitoring response to treatment. For example, the presence of SF3B1 mutation has been included as a diagnostic criterion for MDS with ring sideroblasts in the revised 4th edition WHO Classification.15 Many mutations have been found to be associated with patient outcome and some of the mutations (e.g., EXH2, ASXL1, TP53, RUNX1, and ETV6), have been shown to independently predict clinical behavior independent of other established prognostic variables.20,22 The International Prognostic Scoring System (IPSS), recently revised in 2012 (IPPS-R), has been in use for the past two decades in newly diagnosed MDS patients in order to assess their prognosis and to help in the timing of therapy and to decide among treatment options.18 These scoring systems are based on the degree of cytopenia, bone marrow blast counts and cytogenetic findings. The IPSS-R is considerably more complex than the IPSS, using a larger number of blast strata, five instead of three karyotype categories, and more detailed assessments of cytopenias in each lineage (Table 7.7). The combined data from the above parameters are used to calculate five risk groups (Table 7.8). The IPSS-R has been widely adopted in clinical practice as a basis for treatment decisions and appears to predict outcome both at diagnosis and dynamically in patients being followed at later timepoints.23,24 Although MDS-associated mutations have not been incorporated into the IPSS-R, future prognostic risk systems will likely incorporate the mutational profile in patient risk grouping.
Treatment options for patients with MDS typically fall into one of three broad categories: supportive care (antibiotics and red cell and platelet transfusions for symptomatic anemia and thrombocytopenia), low-intensity therapies (hypomethylating agents, immunosuppressants, and lenalidomide) and high-intensity therapy (multiagent chemotherapy and allogeneic transplant). Although low-intensity therapies are typically used in patients with very low-risk and low-risk IPSS-R scores, high-intensity therapies are usually reserved for high-risk and very high-risk IPSS-R score patients. This lessens treatment-related morbidity and mortality in patients with a relatively good prognosis, and allows for the aggressive treatment of disease in those with a poor prognosis. Patients with intermediate IPSS-R scores may benefit from either of these approaches.25 Lower risk patients with anemia (especially in the context of MDS with ring sideroblasts) and serum erythropoietin (EPO) levels below 500 U/L may benefit from erythropoiesis-stimulating agents used with or without a granulocyte colony-stimulating factor (G-CSF)26; patients with higher serum EPO levels rarely respond to erythropoiesis-stimulating agents.27 Similarly, G-CSF and thrombopoietin receptor agonists may be used in MDS patients with neutropenia and thrombocytopenia, respectively, but none of these agents improve survival.28 The therapeutic approach to MDS will likely increasingly be governed by somatic mutations that can help define responsiveness to particular treatment regimens. For example, certain mutations, such as DMNT3A and TET2, appear to be associated with a superior response to hypomethylating agents. Lenalidomide, a thalidomide analogue, is most effective in MDS with del(5q) lacking TP53 mutation, with a 67% response rate and a median duration of response of more than 2 years.29 Allogeneic transplant, although the only curative modality, is currently used in less than 10% MDS patients, due to advanced age, the presence of comorbidities, and lack of suitable donors. It remains a treatment of choice for children and patients under 40 years of age and is also used in some older patients with higher risk disease. However, the presence of TP53 mutation is associated with poor outcome in MDS treated with allogeneic transplant.30,39
Although establishing an MDS diagnosis relies on the presence of cytopenia(s) and morphologic dysplasia, these features are also present in a variety of neoplastic as well as non-neoplastic conditions (Table 7.9). Some patients have prolonged unexplained cytopenia(s), yet do not meet diagnostic criteria for MDS (due to lack of sufficient dysplasia, increased blasts, or a defining cytogenetic abnormality), a condition designated as idiopathic cytopenia of undetermined significance (ICUS).31 Moreover, MDS-associated mutations may occur in cytopenic or non-cytopenic older individuals who do not have MDS, termed clonal cytopenia of undetermined significance (CCUS) and clonal hematopoiesis
of indeterminate potential (CHIP). Mutation frequency increases with age and individuals with clonal mutations are at a higher risk of development of a hematologic malignancy.32 However, mutations by themselves do not currently establish an MDS diagnosis and in the absence of diagnostic criteria for a myeloid neoplasm, a diagnosis of CCUS/CHIP should be considered.
of indeterminate potential (CHIP). Mutation frequency increases with age and individuals with clonal mutations are at a higher risk of development of a hematologic malignancy.32 However, mutations by themselves do not currently establish an MDS diagnosis and in the absence of diagnostic criteria for a myeloid neoplasm, a diagnosis of CCUS/CHIP should be considered.
Reflecting the recent pivotal developments in the molecular biology of MDS, the terminology and diagnostic criteria of MDS have been updated in the 2016 revision of WHO MDS Classification. The current classification of MDS includes six distinct entities and one provisional entity and is based primarily on bone marrow findings (Table 7.10). Despite the presence of genetic abnormalities in greater than 90% of cases, most of the MDS subtypes are still mainly defined by the degree of dysplasia and blast count, reflecting our incomplete understanding of MDS molecular pathogenesis and challenges in distinguishing MDS from ICUS and CHIP.15 Importantly, certain features exclude a diagnosis of MDS, even in the context of cytopenia and significant morphologic dysplasia (Table 7.11).
MDS WITH SINGLE LINEAGE DYSPLASIA
MDS with single lineage dysplasia (MDS-SLD) is an indolent MDS subtype that accounts for 5% to 10% of MDS cases, with a median survival of about 5 years and acute leukemia development in less than 5% of patients. Dysplasia affects over 10% of cells in only one hematopoietic lineage, ring sideroblasts are rare (<15% of erythroid cells) or absent, blasts are less than 5% of cells in the marrow and less than 1% of cells in the blood, and there is single or bi-cytopenia.15 The most common presentation is with anemia and dysplasia isolated to the erythroid lineage, previously termed refractory anemia in older MDS classifications.14 On the peripheral blood smear the red cells are normocytic or macrocytic. Cytogenetic abnormalities occur in 25% to 50% of cases.
MDS WITH RING SIDEROBLASTS
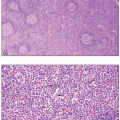
The subtype MDS with ring sideroblasts (MDS-RS) is characterized by ring sideroblasts affecting at least 15% of bone marrow erythroid precursors on an iron-stained bone marrow aspirate smear, or at least 5% ring sideroblasts in the presence of an SF3B1 mutation; blasts must constitute less than 5% of the bone marrow cells and less than 1% of the peripheral blood leukocytes (Fig. 7.13). It is further divided into subtypes with single lineage dysplasia (MDS-RS-SLD), which by definition must be limited to the erythroid lineage and be accompanied by anemia with or without one other cytopenia, and multilineage dysplasia (MDS-RS-MLD).15 The SF3B1 mutation, which affects RNA splicing, is strongly associated with the presence of ring sideroblasts and correlates with a favorable prognosis in MDS.33 Cytogenetic abnormalities occur in less than 10% of cases with single lineage dysplasia, but are more common in cases with multilineage dysplasia. Prognosis in MDS-RS is variable and appears to be poorer in cases with multilineage dysplasia, those that lack SF3B1 mutation, and those that have additional non-SF3B1 mutations such as TP53 and ASXL1.34
MDS WITH MULTILINEAGE DYSPLASIA
MDS with multilineage dysplasia (MDS-MLD) is one of the more common MDS subtypes and the diagnosis requires cytopenia(s) and dysplastic changes (≥10% of cells) in at least two hematopoietic lineages; blasts must comprise less than 5% of the bone marrow cells and less than 1% of the peripheral blood leukocytes. There may be a single cytopenia, two cytopenias, or pancytopenia.15 The median survival is 2.5 to 3 years, and the risk of developing leukemia is approximately 10%. Cytogenetic abnormalities occur in about 50% of cases.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


