Chapter 40 Mycobacterium tuberculosis
All who mix with tuberculosis patients got infected, but remained well so long as they took care of themselves and kept the soil in a condition unfavourable for the growth of the seed.1,2
TB EPIDEMIOLOGY AND CONTROL STRATEGY
Global
Mycobacterium tuberculosis infection is one of the most common infections in the world. Globally, it is estimated that one-third of the world’s population is infected with TB without any evidence of disease. Infection prevalence in the Southeast Asia region is estimated at 44%; Africa, 35%; and Europe, 15%. Individual country rates vary among the highest TB incidence countries: 64% in Cambodia, 49% in Indonesia, 44% in India, 44% in Peru, 38% in South Africa, and 36% each in China, Nigeria, and Kenya.3 Despite an available cure since the 1940s, in 1993 the World Health Organization (WHO) declared TB a global emergency, largely reflecting the neglected worldwide epidemic of undetected and untreated TB cases combined with a lack of international interest or funding for TB treatment and control.4 This declaration followed the establishment in 1991 of a global program framework and case management strategy known as DOTS (Directly Observed Therapy, Short Course). The DOTS strategy is based on five pillars that mix technical and managerial elements, including: laboratory services to diagnose infectious cases; standardized chemotherapy using directly observed therapy; an uninterrupted supply of quality-assured drugs; and a recording/reporting system for monitoring treatment outcomes.5 Over the last several years, the DOTS strategy has expanded to incorporate the challenges inherent to the diagnosis and care of drug-resistant cases and persons dually infected with TB and HIV.6–9
In 2005, WHO declared TB an emergency for Africa.10 For 2004, an estimated 8.9 million persons developed new TB disease worldwide; 1.7 million died from TB, including 229 000 cases co-infected with HIV.11 Among the new cases, some 8.3% were HIV-infected worldwide. However, adult co-infection rates (ages 15–49 years) are substantially higher in many countries: Cambodia, 13%; Ethiopia, 21%; Kenya, 29%; Tanzania, 36%; Mozambique, 49%; South Africa, 61%; and Zimbabwe, 69%. Surveillance data for 2003 showed an overall increase in the global TB incidence of 1% compared with 2002, impacted most strongly by the enormous increase in incidence occurring in sub-Saharan Africa, particularly in high HIV prevalence countries (adult HIV prevalence >4%). Except for the African region, TB incidence either fell or was stable elsewhere.11–13 It is expected that TB prevalence will double in high-HIV areas of Africa to 609 per 100 000 by the year 2015 (compared with 288 in 1990).12
United States
As of 2000, there were an estimated 9.5–14.7 million persons with latent TB infection (LTBI) in the United States, representing 3.4–5.2% of the counted population.14 The US TB case rate dropped an average of 5.5% per year from 1953 to 1985 (range 0–11%). From 1986 through 1992, the number of TB cases and annual US rate increased. This increase was attributed to: deterioration of the public health care infrastructure; emerging HIV epidemic; immigration from countries where TB is highly prevalent; and spread of TB within congregate settings and facilities (homeless shelters, correctional institutions, nursing homes). Since 1992, an annual decline in reported cases resumed. In 2004, there were 14 517 TB cases reported to the CDC, representing a 46% decrease since 1992. However, the number of foreign-born cases reached a new level at 54% of all cases, and for the first time, there were more Hispanic cases than non-Hispanic blacks. The TB case rate for foreign-born persons in 2004 was nearly nine times that of US-born persons (22.8 vs 2.6 per 100 000).15 TB-HIV co-infection data is incomplete nationally due to limited testing and reporting as of 2004.
The principles and practice of TB control in the US are similar to those of the WHO. However, given the available resources and low disease burden, TB ‘elimination’ is the goal with an expanded emphasis on specific control components. The main components are: (1) early identification and treatment of all cases using a rifampin-based regimen, employing directly observed therapy as standard of care; (2) contact investigations around each index case, prioritized by the level of infectiousness (i.e., sputum smear positive, cavity on chest radiograph), contact risk factors (i.e., age, HIV status) and type of exposure environment (open, closed, ventilated); secondary cases are identified in this manner; (3) targeted testing of at-risk groups with treatment of LTBI to reduce the risk of subsequent disease activation; (4) infection control strategies for high-risk settings such as hospitals, health care facilities, prisons and congregate settings such as nursing homes and homeless shelters; and (5) testing of each TB isolate for the presence of drug resistance to ensure adequate therapy is used. Given the limited efficacy of the current BCG vaccination demonstrated in the US, particularly its inability to prevent infection and the limited transmission of TB in the general community, BCG vaccination is not a component of the US strategy.14
MYCOBACTERIUM TUBERCULOSIS AND THE M. TUBERCULOSIS COMPLEX
Mycobacterium tuberculosis is part of what is called the MTB complex, a six-member group of closely related pathogens distinguished by a lack of genetic diversity with a highly stable and conserved genome.16,17 Members of the MTB complex are considered a single species as defined by DNA/DNA hybridization studies.18,19 This complex includes M. tuberculosis (which is the predominate cause of human TB worldwide), M. bovis (which causes TB in cattle, deer, elephants, other wildlife and less frequently in humans since milk pasteurization was introduced), M. africanuum (which is largely limited to sub-Saharan Africa but reported in Europe and recently in the US),20 M. canettii (a smooth, glossy variant of MTB and rare cause of human TB), M. microti (found only in voles or meadow mice) and M. bovis bacille Calmette-Guerin (M. bovis BCG, which is the live attenuated derivative of M. bovis used globally as the vaccine against human TB).
Clinical and Mycobacterial laboratories do not routinely distinguish between these related organisms when reporting a culture as positive growth for TB (meaning M. tuberculosis complex). Discrimination between them is possible, however, through various methods or combination of methods including spoligotyping, DNA fingerprinting using restriction fragment length polymorphism (RFLP) analysis, known deletion mutations, biochemical testing (niacin production, nitrate reduction), susceptibility or resistance to pyrazinamide and morphology.18,20 The doubling time of M. tuberculosis and other members of the complex is ∼22 to 24 h.
Evolution
Except for M. tuberculosis complex and M. leprae, most mycobacteria are commonly found in soil and water and not transmitted from person to person. Today, these so-called ‘environmental’ mycobacteria are collectively grouped as nontuberculous Mycobacteria (NTM) or Mycobacteria Other Than Tuberculosis (MOTT). They include M. avium-intracellulare or M. avium complex, M. kansasii, M. abscessus, M. chelonae and M. ulcerans (Buruli ulcer disease), among several dozen validly described species.21,22 The M. tuberculosis complex likely evolved to infect mammals from similar environmental sources,18 and M. tuberculosis became a human pathogen some 10 000 to 15 000 years ago.23,17 While it was believed that M. tuberculosis evolved from M. bovis (presumably after the domestication of cattle), recent analysis of the evolutionary pathway shows this is not the case. Rather, the M. tuberculosis lineage appears to have separated from a common ancestor before the lineage leading to M. bovis.16
Transmission
The primary means of TB transmission is person to person via the airborne route. Rarely, TB is transmitted at birth, through an open wound or across the skin through direct inoculation of infectious material. Patient-generated aerosols contain a mass of droplets expelled through talking, coughing, sneezing and singing. While most large droplets will fall to the ground quickly, smaller ones may float in the air for a long enough period to allow for droplet evaporation, yielding infectious droplet nuclei. It is these droplet nuclei of 1–5 μm in diameter that find their way to the lung alveoli where infection may be established.24 Transmission is influenced by the concentration of organisms in the air (number of TB organisms/volume of air) and total duration of exposure, which are directly impacted by the type of exposure environment (i.e., ventilation and air exchange), characteristics of the source case (i.e., sputum smear positive for acid-fast bacilli, AFB; presence of a cavity on chest radiograph) and contact characteristics (i.e., relationship to the source case, immune status, genetic susceptibility).25–28 In a molecular epidemiology study of clustered cases in San Francisco, 17% of secondary cases were determined to have resulted from transmission by a smear negative case. The relative transmission rate of a smear negative compared with smear positive source case was 0.22 and not impacted by HIV status.29
PRESENTATION OF TB IN HIV-INFECTED INDIVIDUALS
The clinical manifestations of TB are frequently altered in the setting of HIV co-infection. Typical symptoms and clinical patterns of disease are encountered in those with relatively preserved immune function.30 As immune function deteriorates, there is a progression toward increased adenopathy, involvement of lower and middle lobes of the lung, less cavitation (Fig. 40-1), and more extrapulmonary disease.31–35 Confirmed TB may also be found in those without any symptoms of disease.36 The degree of immunosuppression affects the disease location as demonstrated by a progressively higher frequency of extrapulmonary involvement with advancing CD4 lymphopenia.37 In clinical studies, pulmonary disease has been demonstrated over a wide range of CD4 counts with localized extrapulmonary disease, meningitis, and dissemination occurring with progressively lower median CD4 counts (Fig. 40-2). TB pleural effusions may occur at rates similar to those seen in patients without HIV,38,39 although a study from South Africa demonstrated a significantly higher rate among HIV-positive patients than HIV negative (38% vs 20%).35 Pleural effusions have been documented to occur less frequently in those with CD4 counts <200 cells/mm3 compared to those with CD4 counts >200 (10% vs 28%).37 Further, pulmonary disease often occurs following recent infection as documented by molecular epidemiologic studies showing HIV infection to be associated with clustering, suggesting recent transmission.40–42
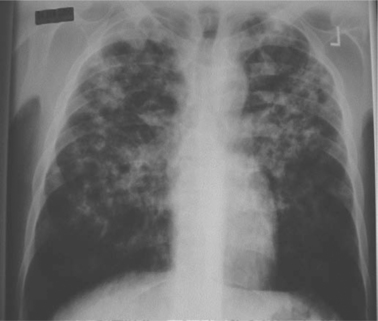

As noted, chest radiograph findings in HIV-infected TB patients vary. Those with more advanced immunosuppression have less cavitation and a higher frequency of lymphadenopathy.31–33 A study from Cote D’Ivoire found cavitary disease in 53% of HIV-infected TB patients with CD4 counts >200 and only 29% in those with more advanced immunosuppression. HIV-negative (control) patients had cavitary disease on 56% of chest radiographs. Additionally, there was incremental progression, with lower rates of cavitation and higher rates of lymphadenopathy, when stratified by decreasing CD4 cell counts.32 The use of highly active antiretroviral therapy (HAART), however, has been associated with more typical CXR findings in HIV-infected patients with pulmonary TB.43 Subclinical TB has also been described in the HIV-infected. A Tanzania study detected TB in 14 of 93 patients screened. Four of these reported no symptoms of TB and had a normal chest radiograph; the diagnosis was made by sputum culture.36
Several clinical studies suggest that HIV-infected patients with culture confirmed pulmonary TB are less likely to be sputum smear-positive than their HIV-negative counterparts31,44–47; however, this is not universally found.48–50 The studies demonstrating no differences in smear-positivity rates utilized fluorescence microscopy. This methodology, when compared with routine Ziehl–Neelsen staining, has been shown to be more sensitive and more likely to detect bacilli in paucibacillary cases51,52 and is not impacted by HIV status.52 Most studies,37,52a,52b but not all,48 have shown an association with lower CD4 cell counts and smear positive sputum status.
In the setting of advanced HIV infection, AFB blood cultures have been shown to be a valuable tool in the diagnosis and confirmation of disseminated TB. M. tuberculosis bacteremia is frequently encountered in febrile AIDS patients in areas of moderate to high endemicity, ranging from 12% to 38%.53–55 The frequency of mycobacteremia correlates with the degree of immunosuppression. In one study of known TB patients in Los Angeles, M. tuberculosis was isolated from the blood of 49% (18 of 37) of patients with CD4 <100, 20% (3 of 15) with CD4 from 101 to 200, 7% (1 of 15) with CD4 from 201 to 300, and in none among eight patients with CD4 >300.37 In two studies from Botswana and Brazil, blood culture was the sole means of diagnosis in 15% of cases.54,56 The Botswana study involved TB suspects with cough. Overall, 22% of the study cohort was found to have a positive blood culture, including several patients also having a positive sputum culture.
EFFECT OF TB/HIV CO-INFECTION AND THE IMPACT OF HAART ON TB
TB has a deleterious effect on the natural history of HIV infection demonstrated by worse clinical outcomes and enhanced viral replication. Marked increases in viral replication are associated with active TB. Goletti et al observed five- to 160-fold increases in viral replication in patients with active disease following the diagnosis of TB.58 TB has also been associated with increased mortality among HIV-infected individuals and may be associated with higher rates of subsequent opportunistic infections (OIs). Two European studies demonstrated significantly higher mortality rates associated with the acquisition of clinical TB, with relative risks of death of 1.34 (95% CI, 1.12-1.60)59 and 1.5 (95% CI, 1.2-2.1).60 A study from Uganda found higher mortality associated with pulmonary TB in HIV-infected patients (RR 1.81, 95% CI, 1.24-1.65). The mortality effect was even more pronounced for those with CD4 counts >200 (RR 3.0, 95% CI, 1.62-5.63),61 when other OIs are less likely to occur. A study in the US comparing HIV-infected subjects with and without TB found higher mortality rates in those with TB. A trend toward a higher frequency of subsequent OIs was also found (RR 1.42, 95% CI, 0.94-2.11),62 suggesting TB may accelerate the time course of HIV infection.
Among TB patients, mortality is consistently higher among HIV-positive than HIV-uninfected individuals.63–65 In a study by Perriens et al of pulmonary TB,65 12 month survival was 69% in the HIV-infected and 98% in the HIV-negative group. TB attributable mortality was 27.8% among those with HIV. Chaisson et al, in a study from Haiti including pulmonary and extrapulmonary disease, demonstrated 33% mortality at 18 months in the HIV-positive group compared with 3% in the HIV-negative group.64 HAART was not utilized in these studies. Diminished CD4 counts less than 100 and not using DOT have been associated with poorer survival among the HIV-TB co-infected.66 A study of HIV-infected, smear-positive pulmonary TB cases from Uganda found anergy to tuberculin, CXR findings atypical for TB, lymphopenia, and a previous diagnosis of an HIV-associated illness all to be independent predictors of death.67 Observational data demonstrates a large proportion of early deaths (<30 days) are attributable to TB,68–70 while a majority of deaths beyond the first month of treatment have been attributed to causes other than TB.69
Impact of HAART on TB
In vitro data has demonstrated improved immunologic responses to tuberculin antigens following the initiation of HAART.71 Such responses remain less robust in HIV-infected individuals on HAART, however, than in HIV-negative controls.72 The improvement in immune response may be limited in those with more advanced immunosuppression prior to TB diagnosis (CD4 <50) despite an adequate response to HAART (a rise in CD4 count of 150 copies/ml from baseline at 12 months and viral load (VL) <50 at 24 weeks).73 These data, when considered together, imply that HAART is not sufficient to restore full anti-TB cellular immune responses.73,74 Nonetheless, HAART has been associated with improved mortality rates in those with active TB.43
Importantly, the use of HAART has been shown to lower the risk of developing active TB with risk reductions ranging from 80% to 92% across studies.75–78 However, a study in South Africa demonstrated a significantly lowered incidence only in those with moderate to advanced immunosuppression, as TB manifests more frequently in those with lower CD4 counts. The incidence rate without HAART was 17.5/100 patient-years for CD4 <200 (3.4 with HAART) and 12.0/100 patient-years for CD4 between 200 and 350 (1.7 with HAART). The incidence risk ratio was not significantly reduced for CD4 values >350, although some cases were averted with HAART use.75 This study excluded pregnant and lactating women.
Despite HAART, TB rates remain higher for those with HIV infection compared to those without HIV infection in both low and high prevalence settings.75,79 In a large cohort analysis of HIV-infected patients across Europe and North America, among those initiated on HAART and followed for 3 years, a low baseline CD4 count, a low CD4 count at 6 months, and a viral load >400 copies/mL after 6 months of HAART were independently associated with a higher risk of developing TB80.
While antiretroviral therapy (ART) is clearly an invaluable component of care for the individually co-infected patient and in combating the global TB epidemic, HAART alone is unlikely to reverse the high rates of TB associated with HIV infection. Extensive modeling of the potential impact of HAART on subsequent development of TB suggests that universal HAART for CD4 <200 would produce only modest reductions in the cumulative incidence of TB (22% over 20 years). Extending HAART to effectively cover 85% of those with a CD4 count up to 500 mL would result in an incidence drop of only 50% over 20 years.81 The less than expected impact reflects the greater life expectancy of those on ART and therefore a longer opportunity to develop active TB, albeit at a lower rate than without ART.81,74 A second model suggests that improving TB diagnosis and cure rates are the most cost-effective means of controlling TB when ART therapy is included in the control strategy.82
DIAGNOSIS OF DISEASE
Before TB can be diagnosed, it must be suspected. There are four steps in diagnosing TB (Fig. 40-3):
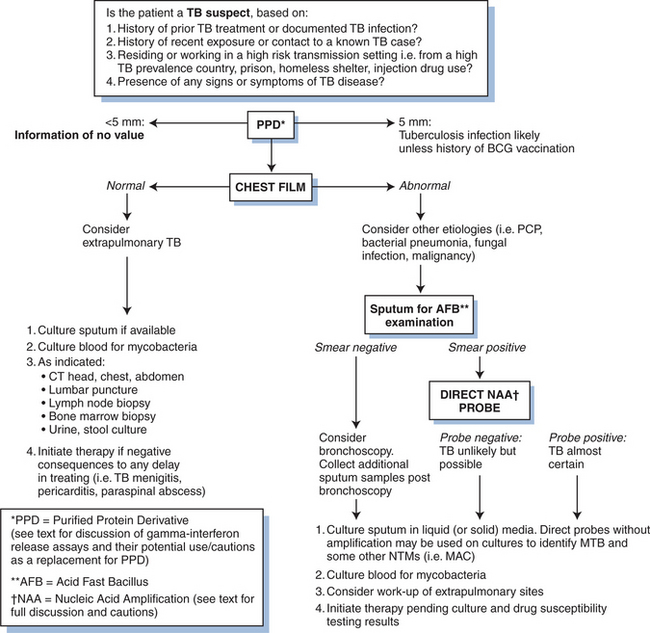
Step 1
Think TB and obtain a good medical history- The initial step is to determine if the patient has been exposed to a person with active disease, has symptoms of TB, a previous history of LTBI or active disease, and risk factors for TB. Exposure may be higher for individuals in certain professions or those living in congregate settings. Persons with TB may be symptomatic or asymptomatic, including HIV co-infected patients.36 If the suspect patient has been treated for LTBI or previous disease, it is important to determine the drugs used and compliance with any therapy.
Step 2
Give the patient a tuberculin skin test (TST) using the purified protein derivative (PPD) of M. tuberculosis. In individuals without confirmation of disease, the TST is the primary means to determine TB infection. It relies upon the delayed-type hypersensitivity (DTH) reaction, which is promoted by interleukin-2 and interferon-gamma. Skin test conversion normally occurs within 8 weeks of infection. However, given the numerous limitations of PPD skin testing (namely cross reactivity with NTM/MOTT and BCG vaccination), recently developed serum-based gamma interferon-based release assays are increasingly available for use in some settings (see further ahead). The currently used TST consists of 0.1 mL of 5 tuberculin unit (5 TU) that is injected intradermally on the volar surface of the forearm using the Mantoux method (1 TU and 250 TU skin tests are uninterpretable with no clinical utility83). The induration at the injection site (not erythema) is recorded in millimeters at 48 to 72 h. Multiple puncture (Tine) PPD skin tests are not reliable,84 and results should be confirmed with a tuberculin PPD test (except when there is blistering at the site of puncture). Up to 20% of patients with active TB who are HIV negative will have a negative PPD skin test, which may convert to positive after initiation or completion of therapy.84–86 Patients with miliary disease are even more likely to have a negative skin test (50–70%).85 Thus, TB should never be ruled out because of a negative PPD. Because of the window period between exposure/infection with MTB and the time to mount a DTH response, repeat PPD testing is recommended 8–10 weeks after contact with the infectious case has been broken.87
Because the TST result must be interpreted in the context of the estimated TB prevalence within an individual’s community and the prior probability of having a true TB infection, various cut-points have been established to determine a positive result.88–90 The cut-point for a positive PPD in an HIV-infected patient is 5 mm or greater, the same cut-point used for a recent contact to a TB case. False positives occur more often in areas with a high prevalence of NTMs and in individuals vaccinated with BCG. False negative reactions may be due to anergy and can occur in very young children or those who are very ill.84 The booster phenomenon results when reactivity to PPD has waned over time and a PPD skin test boosts the cellular immune response. If a second TST is given, the results may be positive and misinterpreted as a new conversion. For this reason, initial two-step skin testing (a repeat TST, 2 weeks after a negative result) is recommended to establish an accurate baseline in individuals who will undergo serial TST testing due to exposure risks.90–93
Anergy Testing and TB
Due to its unreliability and lack of standardization, anergy testing is no longer recommended by the CDC as a routine adjunct for assessing the reaction to the PPD skin test in HIV-infected persons.94 Nonetheless, this issue remains a point of confusion for many physicians. Anergy is the inability to mount a DTH immune response to one, several, or all skin-test antigens to which a recall response is expected due to previous exposure. Anergy therefore may be selective or general.85 Anergy testing is done with common DTH antigens, such as Trichophyton, Candida, histoplasmin, mumps, tetanus toxoid, and tuberculin. Common causes of anergy include: infection with HIV, live viral vaccines, steroids and other immunosuppressive agents, alcoholism, stress such as major trauma, extremes of age, chronic medical conditions, and either disseminated or nondisseminated TB disease.83,85,94,95 People with any of these conditions may have a negative PPD result even if they are clearly infected with MTB. In HIV-infected patients, the likelihood of anergy and PPD nonreactivity are greater when absolute CD4+ T-lymphocyte counts are lower. Anergy is also common in HIV-negative controls (7–23%).96–99
Anergy testing is not useful for interpreting a negative TST result in patients infected with HIV. HIV patients who are anergic to a standard anergy panel at initial testing may respond to these antigens on repeat testing.95,98–100 Many people react to tuberculin but not to the test antigens, or vice versa, and the inability to mount a DTH response to one antigen does not necessarily predict anergy to other antigens.95,96,101,102 Since both tuberculin skin testing and anergy testing are unreliable in HIV-infected persons, these tests should not be used to justify withholding treatment for TB. Further, HIV-infected individuals who are identified as contacts of an infectious pulmonary TB case, and without evidence for disease, should receive treatment for latent TB infection regardless of the TST results, particularly when the likelihood of transmission is deemed high (high proportion of contacts examined with evidence of LTBI) or when a false-negative test is suspected.14,87,103
Gamma Interferon-Based Release Assays to Detect TB Infection
Beyond the concern for a false-negative result in an HIV-infected person, the primary limitation of the TST is its lack of specificity, particularly its inability to discriminate between LTBI and prior BCG vaccination or infection with NTM/MOTT.90 Consequently, serum assays have been developed based on two protein fragments, early secretory antigenic target-6 (ESAT-6) and culture filtrate protein-10 (CFP-10), not found in any BCG vaccine strain nor most MOTT, except for M. kansasii, M. marinum and M. szulgai.103 The CDC issued guidelines in 2005 after FDA approval of one test, QuantiFERON-TB Gold, which uses synthesized proteins. This assay requires fresh heparinized whole blood incubated with the test antigens and measures the supernatant concentration of released interferon-gamma (IFN-γ) by ELISA. Incubation of collected blood with the antigens must occur within 12 h of collection and proper storage of the plasma for ELISA testing must be guaranteed. These requirements can limit the feasibility of the test in multiple field settings. Further, the sensitivity of the test has not been determined for immunocompromised patients (including HIV-infected) and young children. Nonetheless, the CDC has recommended its use in ‘all circumstances in which the TST is currently used’.103 Advantages of the assay are that two visits are not required for placement and reading, as for PPD, and the measurement is automated.
A second IFN-γ release assay uses isolated peripheral blood mononuclear cells rather than whole blood. The test, based on the same antigens ESAT-6 and CFP-10, employs a rapid enzyme-linked immunospot (ELISPOT) assay. Upon mixing, IFN-γ spot-forming T cells are counted. Despite improved specificity, it is not known if the assay is more sensitive than TST for LTBI detection. In a study of Zambian TB patients, the sensitivity of the assay in HIV co-infected TB patients exceeded 90%; no comparison was made with a PPD skin test. Among the study participants without evidence of TB, those who were HIV-infected had a lower median response to the ELISPOT assay than those without HIV infection.104 This finding was attributed to a loss of CD4 T cells and again raises the issue of the accuracy of IFN-γ assays, which are T-cell-based tests, for determining LTBI status in HIV-positive individuals. Nonetheless, the technical performance of one commercial test (T-SPOT TB) has been demonstrated in HIV-infected patients with varying CD4 cell counts, but it has not been prospectively evaluated in the field for TB disease or LTBI.105 In Senegal, while some data indicate that the ELISPOT-based assay is better than standard TST for HIV-infected persons without clinical or radiological evidence of disease (presumed LTBI), there was a significant falloff in the results as CD4 counts fell, especially below 200 cells/mm3.106 In a recent trial directly comparing QuantiFERON-TB Gold with T-SPOT TB, there were a significantly higher proportion of indeterminate results with QuantiFERON-TB Gold (11%) compared with T-SPOT TB (3%). Immunosuppression from cancer chemotherapy was significantly associated with an indeterminate result for both tests, while age <5 years was associated with a greater proportion of indeterminate results using QuantiFERON-TB Gold (32% vs 0%). No HIV data were presented.107
Step 3
Obtain a chest radiograph (CXR). The CXR is useful for determining if there is disease in the chest, and if so, its extent. A CXR, however, cannot confirm or exclude that a patient has TB disease. The problems inherent to CXR interpretation (inter and intraobserver variation) is well described in the TB literature.96,108 CXR is also used to determine if an individual with TB disease is improving on therapy or not. Individuals with HIV co-infection may have active TB and a normal CXR.36 Further, cavity formation is less likely as immunosuppression advances (see section on Presentation of TB in HIV-Infected Individuals). In children, bronchial obstruction may occur from enlarged lymph nodes and may be the only evidence of primary disease, leading to a wedge-shaped segmental or complete lung collapse.
Step 4
Pleural fluid from TB pleuritis is normally exudative with elevated protein concentrations, lymphocyte predominance, and <5% mesothelial cells109 but has relatively low numbers of organisms. Pleural fluid AFB smears are therefore rarely positive, and fluid cultures are positive in only one-third of cases. Pleural biopsy is a more sensitive test and can yield a positive AFB smear, positive culture, or histology showing granulomatous inflammation with areas of caseation necrosis. Pleural biopsy histology or biopsy culture alone can each yield between 60% and 85% of cases, with histology usually a more sensitive test. The combination of histology and culture has been shown to yield ∼95% of TB pleurisy cases.110–112 Given the invasiveness of diagnosis using pleural biopsy and inherent delays awaiting culture confirmation, other means have been sought. High levels of pleural adenosine deaminase (ADA) and IFN-γ are consistently found in TB pleurisy.109,111,113 In a recent study, both tests were demonstrated to be more sensitive for diagnosis (88% and 86%, respectively) than polymerase chain reaction testing (74%). The specificity of ADA and IFN-γ approximated or was better (86% and 97%, respectively) than their sensitivity.113 However in low TB prevalence settings, pleural ADA levels are less specific as it may be elevated with empyemas, lymphomas, rheumatoid effusions and other infections or malignancies.109,112 To overcome this limitation and improve utility, measurement of the isoenzyme ADA2 has been found useful. This isoenzyme is produced only by monocytes and can be assessed by the ratio ADA1/ADApin pleural fluid.112,114,115
Because of the growth characteristics of MTB, traditional solid media culture methods take from 3 to 8 weeks to show growth or not. While commercial broth systems take only 1–3 weeks, neither culture method can readily distinguish between MTB and NTM/MOTT without further testing. Chemical testing, high performance liquid chromatography and nucleic acid probes can distinguish MTB from NTMs. Probes using nucleic acid amplification (NAA) are also available commercially for detecting MTB in clinical specimens directly, but their utility depends upon whether there are a sufficient numbers of organisms present. Consequently, not all NAA products are approved for smear-negative samples, and there is limited information on NAA performance in nonrespiratory specimens. Further, the interpretation of results can be difficult if not confirmed by culture, and interlaboratory variability is seen in test performance (in the sensitivity and specificity). CDC has published a suggested algorithm and cautions for NAA use for TB.116 Not all laboratories have the dedicated resources, personnel and space to routinely use NAA tests, especially on clinical specimens. NAA tests can remain positive after culture conversion to negative during therapy and even after therapy is completed. Therefore, these tests should not be used to monitor treatment and cannot replace core laboratory tests of AFB staining and culture, nor clinical judgment.116
MANAGEMENT OF ACTIVE TB IN HIV-INFECTED PATIENTS
The treatment of TB in patients infected with HIV is similar to that in HIV-uninfected individuals. Recommended treatment regimens and duration of therapy are nearly identical and based on a standard four-drug regimen consisting of 2 months of isoniazid (INH), rifampin (RIF), pyrazinamide (PZA), and ethambutol (EMB), followed by 4 months of INH and RIF (Table 40-1). However, several issues complicate TB management among the HIV-infected. Potential drug–drug interactions exist between antituberculous and certain HAART regimens. Intermittent therapy can also be inappropriate for some patients with HIV co-infection, and the occurrence of paradoxical reactions (PR) and the immune reconstitution inflammatory syndrome (IRIS) can complicate treatment goals.
Table 40-1 Recommended Drug Regimens for Culture-Positive Pulmonary Tuberculosis Caused by Drug-Susceptible Organisms in those Infected with HIV
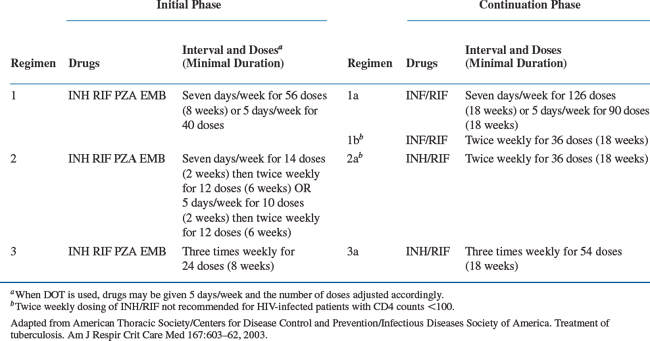
Each agent of the TB regimen has a unique role. INH acts on metabolically active bacilli and is the most potent bactericidal agent. RIF is bactericidal, most effective at sterilization, and helps limit the emergence of drug resistance. PZA is bactericidal against TB bacilli within macrophages in an acidic environment. In combination with INH and RIF during the first 2 months of therapy, PZA is an essential component of the recommended 6-month regimen. EMB is bacteriostatic at doses currently recommended and used primarily to prevent the emergence of resistance. EMB can usually be discontinued after the initial phase of therapy or when drug susceptibility to INH and RIF is verified.117
TB THERAPY ISSUES: DURATION, RELAPSE, REINFECTION AND INTERMITTENT THERAPY
Standard short course (6-month) TB treatment appears to be effective at rapid sterilization and cure among HIV co-infected individuals who complete therapy. Most studies have validated the 6-month regimen as appropriate, with relapse rates less than 5%.63,64,118,119 In a study from Baltimore, Sterling et al found similarly high relapse rates among HIV-infected and uninfected individuals at 6.4% and 5.5%, respectively.120 In a British study of HIV-TB co-infected patients, where HAART was utilized, the 2 year relapse rate was 4.1% at 2 years.121 These findings are not universal, however. Two studies demonstrated higher relapse rates in HIV infected individuals who received 6 months of therapy in comparison with longer courses of treatment.65,122 A study from the Democratic Republic of Congo (formerly Zaire) demonstrated a higher relapse rate at 24 months from enrollment in HIV patients treated for 6 months compared to 12 months, 9% and 1.9% respectively (p < 0.01), but without any survival benefit. Certain limitations, however, apply to this study. DOT was not universally implemented; twice-weekly therapy was utilized in the continuation phase, and the role of reinfection versus relapse was not assessed. Further, the duration of follow-up after completion of therapy differed between the two groups.65 Another study from Spain demonstrated a statistically significant higher relapse rate in those receiving less than 9 months of treatment compared to those receiving more than 9 months, 24% versus 3.4% respectively (p < 0.001).122 In a third study from the US comparing 6 months with 9 months of treatment (all pan-susceptible cases), relapse rates were 3.9% versus 2% respectively, a nonstatistically significant difference.119
Reinfection has been well described in areas of high endemicity. In a study of South African gold miners, 93% (13/14) of recurrences within the first 6 months of completing anti-TB therapy were attributable to relapse. Beyond 6 months, 52% (13/25) were attributed to reinfection. HIV-infection was found to be a risk factor for reinfection but not relapse.123 A separate South African study from an urban population demonstrated that among those successfully completing anti-TB therapy, 77% of subsequent episodes of TB were due to reinfection rather than relapse. HIV status was not determined in this study, however.124
Recently published American Thoracic Society (ATS)/Centers for Disease Control and Prevention (CDC)/Infectious Diseases Society of America (IDSA) consensus guidelines recommend standard 6-month anti-TB treatment regimens for those co-infected with HIV and TB (Table 40-1; see Table 40-2 for drug dosing).125 However, patients with suboptimal responses, defined as sputum culture positive at the end of2 months therapy, should be considered for a 3-month extension of the continuation phase, for a total of 9 months (Fig. 40-4). Further, all forms of TB should be treated based on the same 6-month regimen, except for TB meningitis for which 9–12 months of therapy are recommended. The American Academy of Pediatrics suggests a 9-month treatment regimen for pulmonary TB in all children co-infected with HIV126; 9–12 month regimens are appropriate for children with disseminated or meningeal forms of TB given the lack of specific data to support shorter treatment durations. All HIV-positive patients with TB should be treated with DOT,125 which is especially important when any intermittent dosing regimen is applied. Short course regimens that utilize RIF throughout treatment should be utilized at all possible times as treatment outcomes are superior to those achieved by other combinations as demonstrated by higher treatment failure and relapse rates.127 or increased mortality and delayed sterilization.128
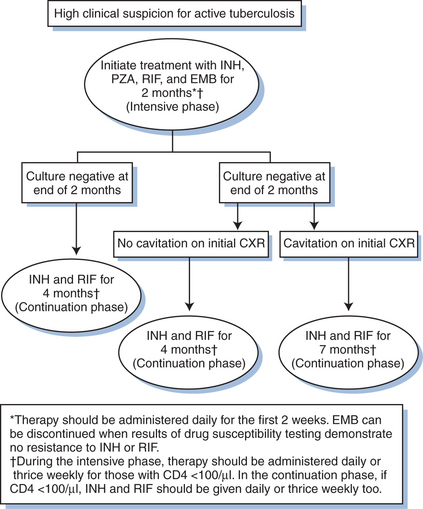
Figure 40-4 Treatment algorithm for pulmonary tuberculosis among those with HIV infection.
Adapted from American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America. Treatment of tuberculosis. Am J Respir Crit Care Med 167:603–62, 2003.
Evidence suggests that intermittent dosing regimens administered less than three times a week may result in poor outcomes among certain HIV-infected groups. Acquired rifampin monoresistance has been seen in patients on twice-weekly dosing regimens.119,129 Acquired resistance has also been observed with twice-weekly rifabutin-based regimens.130,131 Of note, all such cases had CD4 counts less than 100. Weekly dosing of rifapentine, a long acting rifamycin with a half-life five times that of rifampin, is also contraindicated in TB-HIV co-infection. A randomized trial of HIV-related TB comparing weekly INH-rifapentine with twice-weekly INH-RIF use in the continuation phase found a higher rate of relapse in the former group, 17% (5/30) and 10% (3/31) respectively. Four of the five relapses in the INH-rifapentine group and none of the three in the INH-RIF group acquired rifampin resistance.132 Consequently, ATS/CDC/IDSA guidelines recommend avoiding twice-weekly intermittent therapy in anyone with CD4 <100. Furthermore, rifapentine-based continuation phase regimens are contraindicated in those co-infected with TB-HIV, regardless of CD4 levels.125
ADMINISTRATION OF HAART DURING TB TREATMENT
Given the complexities of co-administering HAART with anti-TB therapy, the current consensus is to delay HAART until several weeks of anti-TB therapy have been completed.125 There are no clinical trials defining the optimal time to initiateART, and standard guidelines are based on the underlying degree of immunosuppression. Initiating HAART without delay in those with advanced immunosuppression may be beneficial as they have a high risk of subsequent OIs and OI-related death soon after the initiation of TB therapy. A retrospective study from London comparing outcomes in HIV co-infected TB patients during the pre-HAART and HAART eras demonstrated a high adverse event risk, defined as death or subsequent AIDS defining illness, within the first 2 months of TB therapy. These occurred at a rate of 139/100 person-years pre-HAART and 88/100 person-years post-HAART. Those with advanced HIV disease (CD4 <100) had a higher adverse event rate (249/100 persons-years) in the first 2 months of TB therapy. Paradoxical reactions were common in those with low CD4 counts, most often occurring 15–30 days after HAART initiation; however, the relationship to the timing of anti-TB therapy is not clear.133
Joint ATS/CDC/IDSA guidelines suggest initiating HAART 4–8 weeks after starting TB therapy.125 This delay may simplify adherence issues during the initial phase of TB treatment and reduce confusion if adverse events occur. Adverse medication events, such as peripheral neuropathy, nausea and vomiting, rash, and hepatitis are commonly encountered in patients co-infected with HIV and TB. Of 183 patients studied by Dean et al, 34% had adverse reactions necessitating the discontinuation or cessation of some component of TB or HIV therapy.121 These events are not typically specific to any drug (Table 40-3), and temporal relationships are often the only clues to which agent may be responsible. An additional advantage in delaying HAART may be to diminish the risk of IRIS (see section on Paradoxical Reactions and the IRIS). According to WHO guidelines, patients with CD4 counts <200 should receive HAART as soon as TB therapy is tolerated, which can be between 2 and 8 weeks. For those with CD4 counts from 200 to 350, HAART can be delayed until the initial 8 weeks of anti-TB therapy is completed. For those with CD4 counts >350, deferment of HAART is recommended (Table 40-4).134 Those on HAART prior to the diagnosis of TB should continue taking ART, although dosing adjustments or drug substitutions may be required because of drug interactions.
Table 40-3 Drug and/or Disease-Related Adverse Events
| Adverse Event | Commonly Associated TB and HIV Drugs | Other Possible Etiologies Associated with HIV Disease |
|---|---|---|
| Hepatitis | PZA, INH, RIF, RFB, nevirapine, PIs, trimethoprim–sulfamethoxazole, PAS, ethionamide | Viral hepatitis, IRIS, CMV, EBV |
| Skin eruptions | PZA, INH, RIF, RFB, nevirapine, efavirenz, abacavir, trimethoprim–sulfamethoxazole, streptomycin | Eosinophilic folliculitis, scabies, psoriasis vulgaris, atopic dermatitis |
| Nausea and vomiting/or diarrhea | PZA, INH, RIF, RFB, AZT, ritonavir, amprenavir, indinavir, trimethoprim–sulfamethoxazole, PAS, ethionamide | Pancreatitis, HIV associated cholangiopathy, IRIS, CMV, cryptosporidiosis |
| Hematologic abnormalities (leukopenia, anemia, thrombocytopenia) | RIF, RFB, AZT, valgancyclovir, trimethoprim–sulfamethoxazole, INH (rare) | HIV related bone marrow suppression, ITP, autoimmune hemolytic anemia |
| Visual disturbances | EMB, RFB, voriconazole | CMV retinitis, IRIS, toxoplasmosis, VZV, fungal infections |
| Neuropathy | INH, DDC, ddI, d4T, ethionamide, dapsone | HIV-related, alcohol, CMV |
| Seizures | INH, cycloserine, fluoroquinolones, efavirenz | CNS lesions (lymphoma, toxoplasmosis, PML), meninigitis, HIV-associated |
| Flu-like syndrome | RIF (especially with intermittent therapy) efavirenz, cycloserine | Acute HIV infection |
| Sleep and Psychiatric disturbances |

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


