Fig. 11.1
Molecular landscape of lung adenocarcinoma. Estimation of mutation prevalence is based on targeted next generation sequencing of lung adenocarcinomas performed at Brigham and Women’s Hospital and Dana–Farber Cancer Institute (“Oncopanel”) from 2013–2017. Prevalence data for fusions is based on Awad MM et al. 2016 10.1200/JCO.2015.63.4600 [16]. Please see text for relevant discussion of alterations
Molecular diagnostic techniques, including PCR and single-gene sequencing, fluorescence in situ hybridization (FISH), immunohistochemistry, and, most recently, next-generation (massively parallel) sequencing, have come to the fore in the pathologic characterization of lung adenocarcinoma, and molecular pathologists now play a key role in the multidisciplinary approach to the evaluation of this tumor type. Here we will review the most common targetable mutations in lung adenocarcinoma, the parallel development of molecularly targeted therapies, and the specific molecularly defined resistance mechanisms arising in the face of these therapies.
EGFR
EGFR is a member of the ErbB transmembrane growth factor receptor family and is comprised of an extracellular ligand-binding region, a single hydrophobic transmembrane bridge connected to an intracellular juxtamembrane (JM) region, a tyrosine kinase domain, and an intracellular C-terminal tail with multiple tyrosine residues. Upon extracellular ligand binding, EGFR homodimerizes or heterodimerizes with other members of the ErbB family and autophosphorylates its C-terminal tyrosines, causing downstream intracellular activation of PI3K, JAK/STAT, and MAPK signaling pathways, ultimately leading to cellular proliferation. Approximately 20% of lung adenocarcinomas (Fig. 11.1) harbor somatic activating mutations in EGFR; EGFR mutations more commonly occur in female patients, never-smokers, and patients of East Asian descent [3]. EGFR mutations cluster in the tyrosine kinase domain encoded by exons 18, 19, 20, and 21. The most common activating EGFR mutations (~95%) are a point substitution in exon 21 (L858R) and in-frame deletions in exon 19 involving the LREA motif (Fig. 11.2), which result in constitutive ligand-independent kinase activity [4]. The therapeutic importance of these molecular alterations was shown in the mid-2000s when it was found that lung adenocarcinomas specifically harboring these activating mutations responded to treatment with small-molecule EGFR inhibitors, such as erlotinib, gefitinib, and afatinib, whereas EGFR wild-type tumors did not (reviewed in [4]). This led to a paradigm shift in the diagnosis and treatment of lung adenocarcinoma wherein detection of these genetic alterations was used to select patients for targeted therapy, beginning the era of “precision medicine” in solid tumor oncology.
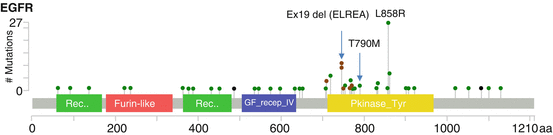
Fig. 11.2
Distribution of mutations in EGFR lung adenocarcinoma. Missense, in-frame deletion, and nonsense mutations are represented by green, brown, and black lollipop symbols, respectively. Adapted from http://www.cbioportal.org (accessed 9 Apr 2017)
Acquired resistance to EGFR-targeted therapy invariably arises in patients at a median of 12 months following initiation of therapy [5]. The most common mechanism (~50%) is the T790M exon 20 mutation (see Fig. 11.2) in the ATP-binding site of the kinase domain, which results in the normalization of increased ATP affinity enabled by the activating EGFR mutation (e.g., L858R) to near wild-type levels [6]. Recently, third-generation EGFR inhibitors (e.g., osimertinib) designed to overcome the T790M-mediated resistance have been approved by the FDA based on reported 95% clinical efficacy of these drugs in phase I clinical trials of patients with EGFR T790M mutations [7]. Phase 3 randomized trials have confirmed the superiority of osimertinib over platinum-based chemotherapy in the relapse setting in terms of disease response, progression-free survival, and adverse events [8]. Thus, detection of the T790M mutation at the time of relapse has become critical to guide further targeted therapy. Non-T790M mechanisms of EGFR resistance also exist, including MET and ERBB2 (HER2/neu) amplification, PIK3CA mutation, and evolution to small cell carcinoma [9], some of which may be amenable to targeted therapy where such agents exist (e.g., MET, ERBB2 inhibitors [10, 11]).
Testing for EGFR mutations is recommended for all patients with lung adenocarcinoma at time of diagnosis ([3]-guidelines update, in preparation) and is routinely performed using a variety of methods, including Sanger sequencing with and without mutated allele enrichment, the amplification refractory mutation system, fragment length analysis, restriction fragment length polymorphism, real-time PCR, and digital droplet PCR. More recently, EGFR mutation testing has been incorporated into next-generation panel-based sequencing platforms allowing for co-detection of potentially targetable molecular alterations and resistance mechanisms. Furthermore, plasma-based PCR testing for circulating cell-free DNA (cfDNA) can detect T790M in circulating tumor DNA from EFGR tyrosine kinase inhibitor (TKI)-resistant patients and is associated with clinical response to osimertinib [12]. Assays using next-generation sequencing of cfDNA are currently widely available; however, the clinical performance of this approach to tumor genotyping in different settings is still under investigation.
MET Exon 14 Skipping
c-Met encodes MET, which belongs to a family of receptor tyrosine kinases that play a crucial role in cell growth and proliferation, survival and apoptosis, epithelial-mesenchymal transition (EMT) and invasion, tissue remodeling, and morphogenesis. MET is physiologically activated by the binding of its specific ligand, hepatocyte growth factor (HGF), leading to the downstream activation of PI3K, STAT, and MAPK pathways [13]. The intracellular MET juxtamembrane domain is partially encoded by exon 14 and contains critical regulatory elements, including a tyrosine at position 1003 (Y1003), which is the direct binding site for Cbl, an E3 ubiquitin ligase that promotes MET protein degradation [14]. Somatic substitution and small deletion mutations at the exon 14 splice acceptor and donor sites leading to exon 14 skipping, as well as rare missense substitutions involving Y1003 (Fig. 11.3), lead to increased stability and oncogenic potential of the MET receptor ([15]). MET exon 14 skipping mutations are seen in 3–4% of non-squamous lung carcinomas (Fig. 11.1), the majority of which are adenocarcinomas. Stage IV tumors with MET exon 14 skipping mutations tend also to show concurrent MET copy number amplification and strong protein expression by immunohistochemistry. MET exon 14 skipping mutations are associated with older age and female gender and are enriched in tumors with a poorly differentiated (see Fig. 11.4) or pleomorphic histomorphology [16, 17]. Isolated case series have shown partial responses of MET exon 14 skipping mutant lung adenocarcinomas to the multitargeted tyrosine kinase inhibitors, crizotinib and cabozantinib [16, 18]. Resistance mechanisms related to MET inhibitors specifically in the context of exon 14 skipping have not been well described; however, kinase domain mutations such as MET p.D1228V that interfere with inhibitor binding have been demonstrated to confer resistance to MET inhibitors when applied in the context of MET gene amplification [19].
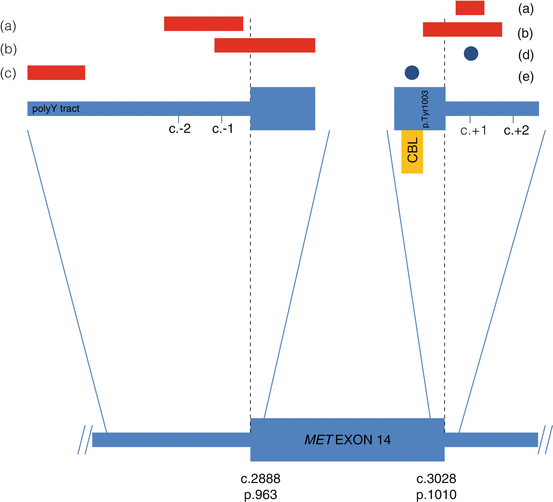
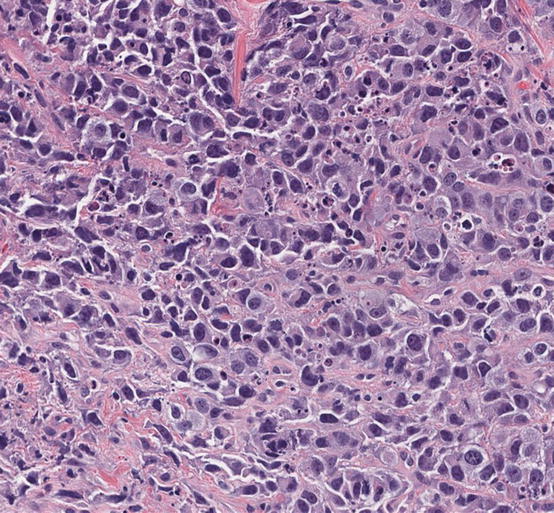
Fig. 11.3
–>Schematic of mutations in MET (NM_001127500.2) that result in exon 14 skipping in lung adenocarcinoma. Black dotted lines represent intron/exon boundaries. Deletions are shown as red rectangles and point mutations are shown as blue circles. Deletions can occur at the (a) canonical splice donor and acceptor sites, (b) span the intron/exon boundaries, or involve the (c) polypyrimidine (polyY) tract in intron 13. Point mutations can occur at the (d) splice donor site or at a (e) key tyrosine residue (p.Tyr1003) that mediates CBL binding
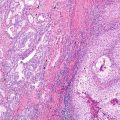
Fig. 11.4
–>Morphology of lung adenocarcinoma with MET exon 14 skipping mutation. This lung adenocarcinoma shows poorly differentiated features, which is often associated with MET exon 14 skipping (and gene amplification). Next-generation sequencing revealed a canonical exon 14 skipping mutation, MET p.D1010N (c.3028G>A), and gene amplification (hematoxylin and eosin staining, 200× magnification)
BRAF
BRAF encodes a serine/threonine kinase that is a key member of the RAS/RAF/MAPK growth and proliferation signaling pathway. While activating mutations of BRAF are very common in certain tumor types (e.g., cutaneous melanoma), BRAF mutations overall are relatively rare in lung adenocarcinoma (~4%, Fig. 11.1). The activating V600E mutation in exon 15 predominates in other tumor types, whereas only about half of lung adenocarcinomas harbor this mutation. About half of the BRAF mutations seen in lung adenocarcinoma occur at other codons in exon 15 or at several hotspots in exon 11 (Fig. 11.5) [20]. BRAF V600E mutations are generally present in a mutually exclusive fashion with other oncogenic drivers, but recurrent non-V600 are more likely to be observed as co-mutations, including with EGFR and KRAS [21]. In contrast to the success of other molecularly targeted therapies, stand-alone BRAF-specific inhibitors have had generally disappointing results in lung adenocarcinoma, with resistance developing within 6 months despite significant initial responses (reviewed in [22]). Resistance mechanisms include constitutive autocrine signaling through EGFR or loss of the full-length BRAF V600E-mutated protein. However, phase 2 trials of the BRAF inhibitor dabrafenib together with the MEK inhibitor trametinib have demonstrated a 63% response rate in patients with BRAF V600E-mutated lung adenocarcinoma with a median duration of response of 9 months [23]; therefore, targeted therapies do hold promise in this setting. Current clinical guidelines do not recommend up-front, stand-alone testing for BRAF mutations, but molecular labs should incorporate testing of the BRAF gene, ideally to include both exons 11 and 15, into multiplex genotyping or next-generation sequencing panel testing [guidelines in preparation].
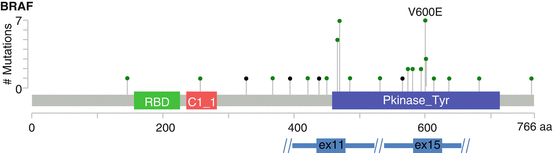
Fig. 11.5
–>Distribution of mutations in BRAF in lung adenocarcinoma. Missense and nonsense mutations are represented bygreen and black lollipop symbols, respectively. Adapted from http://www.cbioportal.org (accessed 9 Apr 2017)
ERBB2
Like EGFR, ERBB2 , also known as HER2/neu , is a member of the ErbB family of growth factor receptors and a well-recognized oncogene in a variety of tumor types. In breast and gastrointestinal carcinomas, ERBB2 amplification is relatively common, leading to activation of downstream MEK and AKT pathways and predicting response to targeted therapies such as trastuzumab and lapatinib. The predominant mechanism of ERBB2 activation in lung takes the form of mutations, rather than amplification. The most common activating mutations of ERBB2 in lung adenocarcinoma occur as small exon 20 in-frame insertion mutations occurring at or around codon 775 in the kinase domain and leading to duplications of some or all of the YVMA amino acid sequence [24]. These mutations lead to constitutive phosphorylation and activation of HER2 and downstream signaling pathways (AKT, MAPK; [25]). ERBB2 exon 20 mutations have been reported in approximately 2% of lung adenocarcinomas (Fig. 11.1) and tend to occur in female nonsmokers and patients of East Asian ethnicity, similar to the characteristics of patients with EGFR mutations [26–28]. Several case studies have shown responses in patients harboring ERBB2-mutant lung adenocarcinomas to ERBB2-inhibitors in combination therapies [29–31]. The largest study to date examining the efficacy of ERBB2-targeted agents in this tumor type is a retrospective study that included a total of 101 patients with ERBB2-mutated lung adenocarcinoma, of which 65 patients received ERBB2-directed therapies with conventional platinum-based chemotherapy. ERBB2-targeted therapies (trastuzumab and trastuzumab-emtansine) in combination with chemotherapy were associated with a 51% overall response rate and 4.8-month median progression-free survival, which was similar to patients receiving chemotherapy alone in the first line [32]. Together, these data suggest that ERBB2-mutant lung adenocarcinoma may be sensitive to targeted therapy, though larger prospective studies are needed to determine true clinical efficacy. Studies are conflicting on the association between ERBB2 activating mutations and copy number changes in lung adenocarcinoma. Limited studies examining ERBB2-mutated tumors suggest a high frequency of concomitant ERBB2 gene polysomy or amplification [33]. However, other studies show that, overall, ERBB2 amplification is rare in lung adenocarcinoma and may co-occur with other oncogenic driver alterations. Further, these studies show that ERBB2 amplification and mutation are largely nonoverlapping, suggesting that these represent distinct mechanisms of oncogene activation and tumorigenesis in the lung [34]. Overall, the number of cases analyzed to date limits the conclusions that can be drawn on this topic. There is little evidence to support routine testing for ERBB2 amplification; however, ERBB2 exon 20 mutation detection should be incorporated into genotyping or sequencing panels for lung adenocarcinoma [CAP/IASLC/AMP Lung Cancer Testing Guidelines, in preparation].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


