Introduction
Myopathies may be inherited or acquired. Although the latter are more common in geriatric practice, several inherited myopathies may present for the first time in middle age or later and may raise genetic issues for other family members. The acquired myopathies, with a few notable exceptions, often recover with appropriate management. For all of the conditions that are discussed in this chapter, appropriate support, even in the absence of specific therapy, will help improve quality of life and reduce morbidity.
The clinical features of myopathies are relatively limited and include weakness and muscle wasting, fatigue, pain, tenderness, myotonia and muscle twitching. At the bedside it may be impossible to decide whether weakness and wasting are due to a myopathy or a neurogenic disorder. Thus, the differential diagnosis of myopathy includes anterior horn cell disorders (e.g. amyotrophic lateral sclerosis and spinal muscular atrophy), nerve root and plexus disorders, peripheral neuropathies and myasthenic disorders. The presence of upper motor neurone signs and sensory symptoms and signs clearly suggests a more proximal cause of problems than skeletal muscle, but in their absence it is important not to forget that weakness alone is often due to nerve rather than muscle disease. The role of laboratory investigations in helping to distinguish between neurogenic and myopathic diseases is discussed below.
There are several ways of approaching the classification of muscle disorders. For example, inherited disorders can be classified by phenotype or by gene/protein product. For everyday usage, a subdivision into acquired and inherited disorders is useful (Tables 67.1 and 67.2). In the elderly (Table 67.3), the most frequently encountered disorders are the idiopathic inflammatory myopathies, drug-induced myopathies and muscle disease in association with endocrine and metabolic disease.
Table 67.1 Acquired myopathies.
Idiopathic inflammatory myopathies
|
Toxic
|
Endocrine and metabolic
|
Infection
|
| Paraneoplastic myopathies |
Table 67.2 Inherited myopathies.
Muscular dystrophies
|
| Myotonic dystrophies |
Congenital myopathies
|
Metabolic myopathies
|
Channelopathies
|
Table 67.3 Major myopathies in the elderly.
| Idiopathic inflammatory myopathies |
| Toxic myopathies |
| Endocrine and metabolic myopathies |
| Oculopharyngeal muscular dystrophy |
| Mitochondrial chronic progressive external ophthalmoplegia |
Clinical Assessment
The patient’s history is likely to be more revealing than physical examination, although laboratory investigations may be required to establish the precise diagnosis.1, 2 In the history, particular attention must be paid to determining the site of onset and rate of progression of skeletal muscle involvement and the presence of associated symptoms such as muscle wasting and pain. There must be a detailed recording of the family history and drug history. For myopathies secondary to systemic disease (Table 67.1), the underlying cause may be identified from the history and physical examination. In practice, the most frequently encountered pattern of skeletal muscle involvement is that of painless proximal weakness affecting the pelvic girdle
more than the shoulder girdle. In the acquired myopathies (Table 67.1), onset may be acute or chronic, whereas in the inherited myopathies (Table 67.2), progression is usually slow. Asymmetric involvement is uncommon but may be a striking feature in inclusion body myositis (IBM) and facioscapulohumeral (FSH) muscular dystrophy. Early involvement of distal muscles is seen in myotonic dystrophy and IBM.
There is early involvement of the extraocular muscles (causing ptosis and/or ophthalmoplegia) in myasthenia gravis, mitochondrial cytopathies, Graves’ ophthalmopathy and oculopharyngeal muscular dystrophy. Ptosis and facial weakness are seen in myotonic dystrophy and facial weakness is a particular feature of FSH muscular dystrophy and some of the congenital myopathies. Myasthenia gravis most frequently presents with intermittent diplopia and ptosis. Subsequently there may be facial and bulbar muscle involvement.
Weak muscles eventually become wasted, but in many myopathies the bulk is maintained for a long time, whereas in neuropathies wasting is generally an earlier feature. The tendon reflexes also tend to be lost early in neuropathies. Muscle hypertrophy is rare in acquired myopathies but is a feature of some of the muscular dystrophies (e.g. Duchenne/Becker dystrophy).
Respiratory muscle weakness may be asymptomatic, but evidence of it can be found on examination in the form of paradoxical abdominal movement. The best method of evaluation is by measurement of the forced vital capacity (FVC). In the presence of diaphragmatic weakness, the FVC falls when the patient lies down.
Most myopathies are painless at rest. Acute dermatomyositis (DM) may be painful, but rarely markedly so, and there is a striking difference between the modest pain and severe weakness seen in this condition and the severe pain and stiffness and absence of weakness seen in polymyalgia rheumatica. Aching is a common feature in hypothyroid myopathy and bone pain a major feature of osteomalacia. Acute drug-induced and acute alcoholic myopathies are often painful. Exercise-induced muscle pain is seen in several metabolic myopathies1 and in Becker muscular dystrophy.
Clinical features alone usually provide a strong pointer toward the diagnosis and help determine the path of laboratory assessment.3
Laboratory Investigations
A fairly common pathway from the clinical evaluation to diagnosis is via biochemical studies, electrophysiological assessment and muscle biopsy. For the inherited myopathies, this pathway is gradually being supplanted in part or in whole by specific DNA tests.3
Biochemical Studies
Despite its lack of specificity, estimation of the serum creatine kinase (CK) is a useful marker for muscle disease. High levels are seen in Duchenne and Becker muscular dystrophy, acute drug-induced and toxic myopathies, inflammatory myopathies, some forms of limb-girdle muscular dystrophy and some metabolic myopathies. Levels are normal in many congenital myopathies, myotonic disorders, chronic drug-induced myopathies and many metabolic myopathies. Moderate elevation (up to 1000 i.u. l−1) may be seen in anterior horn cell disorders. The serum aspartate aminotransferase (AST) level generally parallels the CK level in muscle disorders and not infrequently its elevation in the presence of otherwise normal liver function tests is the first indication of a myopathic disorder—however, this may not be appreciated and some patients undergo unnecessary liver investigations, including biopsy, before the penny drops and the CK is measured.
Dynamic studies (such as estimation of lactate generation during exercise and magnetic resonance spectroscopy) are of value in the investigation of suspected metabolic myopathies involving carbohydrate and mitochondrial metabolism and tandem mass spectrometry is of value in evaluating disorders of fatty acid β-oxidation.1
Electrophysiology
In primary disorders of the muscle, electromyography (EMG) characteristically shows a reduction in the size and duration of the motor unit potentials and an increase in the number of polyphasic units. Fibrillation potentials and positive sharp waves indicate muscle fibre irritability and are seen in inflammatory myopathies and some metabolic myopathies. Myotonic discharges are seen in myotonic dystrophy and myotonia congenita.
EMG may help in distinguishing between neurogenic and myopathic disorders, which may sometimes be difficult on clinical grounds alone.
Muscle Biopsy
With the exception of DNA tests, muscle biopsy remains the single most powerful tool for the specific diagnosis of myopathies.4 The muscle to be biopsied must be chosen with considerable care.3 Tissue handling is extremely important and correct processing requires specialist facilities.1 Biopsy findings in individual disorders are discussed throughout this chapter. Biopsy samples are also used for biochemical studies and mitochondrial DNA analysis.
Molecular Studies
There has been rapid progress recently in the identification of the genetic basis of many of the inherited myopathies (see Gene Tables at http://www.musclegenetable.org/). This offers the prospect of rapid and precise diagnosis and the option of prenatal diagnosis. However, a number of practical difficulties remain in moving tests from the research arena to accredited diagnostic laboratories.5
Acquired Myopathies
The acquired myopathies (Table 67.1) are of particular importance because many of them are either treatable or reversible. The idiopathic inflammatory myopathies are the subject of considerable research activity, which has aided classification and our understanding of pathogenetic mechanisms and is starting to help in determining therapeutic approaches. Toxic and drug-induced myopathies are seen at all ages but are particularly important in the elderly because of polypharmacy. Myopathy, occasionally severe, is common in many endocrine disorders and usually responds rapidly to correction of the underlying condition. Disorders of calcium and vitamin D metabolism in the elderly are an important cause of muscle weakness. Several forms of paraneoplastic myopathy exist but, in general, the condition is overdiagnosed.
Idiopathic Inflammatory Myopathies
Dermatomyositis (DM) and polymyositis (PM) share many clinical features and there are common approaches to treatment, but recent studies have shown that they have very different immunopathogenic mechanisms.6 Inclusion body myositis may not be a true primary inflammatory myopathy but it is often confused clinically with PM and is particularly important in the present context because it is seen most frequently in the elderly.
Myositis may be seen in association with connective tissue disorders (including systemic lupus, scleroderma, Sjögren syndrome, rheumatoid arthritis and mixed connective tissue disease) and these are sometimes referred to as overlap syndromes.6 Care must be taken to distinguish a true inflammatory myopathy as the cause of weakness in these conditions from other associated causes, such as peripheral neuropathy, muscle ischaemia, cachexia and drug-induced myopathy. Overall, clinically relevant inflammatory myopathy in these overlap syndromes is uncommon.
Dermatomyositis (DM) (see Chapter 125, Skin Disorders)
Clinical Features
DM can develop at any age, is twice as common in females than males and has an annual incidence of about two per million population. Onset of weakness is usually subacute (weeks) but, in rare cases, has a more explosive onset with profound weakness, respiratory muscle involvement and rhabdomyolysis with myoglobinuria developing within a few days. Exercise-induced muscle pain and discomfort on palpation may be present but severe discomfort of the type associated with polymyalgia rheumatica is not seen. Dysphagia is common.
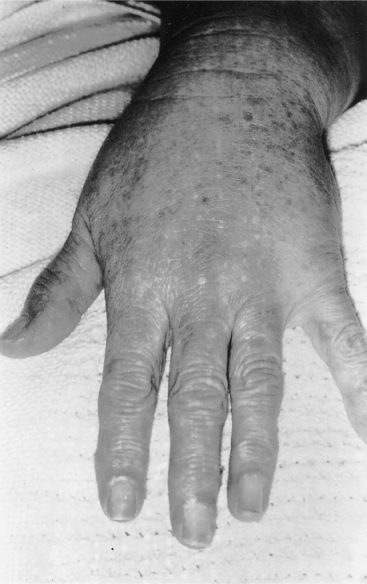
Clinical evidence of skin involvement is present in most, but not all, cases. The commonest features are a non-specific erythematous rash over the face and upper anterior chest wall (areas exposed to sunlight), a red–purple discoloration over the knuckles and dilatation of capillaries at the base of the nail bed (Figure 67.1). Characteristic, but seen less often, is a violaceous (heliotrope) discoloration of the eyelids. Raynaud’s phenomenon, which may long predate the myopathy and arthralgia, is frequent. Interstitial pulmonary fibrosis may be asymptomatic, cause breathlessness and cough or occasionally be relentlessly progressive despite treatment and lead to death. There is an association between lung involvement and the presence of serum anti-Jo-1 antibodies. Cardiomyopathy and rhythm disturbances are underestimated and may also lead to death. In about 20% of cases overall, but more frequently in older patients, there is an association with carcinoma, but not with a particular carcinoma. Thorough clinical assessment is therefore essential, including rectal, vaginal and breast examination, sigmoidoscopy or more extensive endoscopy if there is a suggestive history, abdominal and chest scanning, testing for faecal occult blood and basic haematological and biochemical studies.7 Positron emission tomography (PET) is becoming increasingly available as a screening tool to detect occult malignancy.
Figure 67.1 Dermatomyositis. Note erythema over knuckles and dilatation of the nail-bed capillaries.
Diagnosis and Pathological Features
The serum CK is usually elevated and in acute cases the level may be very high. It is a useful but not absolute indicator of disease activity. The erythrocyte sedimentation rate (ESR) is normal in the majority of patients. Autoantibodies are frequently found but their identification is of little value with respect to diagnosis or prognosis, except perhaps for the association noted above between anti-Jo-1 and interstitial lung disease.8 EMG shows an alteration in the motor unit potentials (reduced duration and an increase in polyphasic units) and electrical irritability (fibrillation potentials and positive sharp waves).
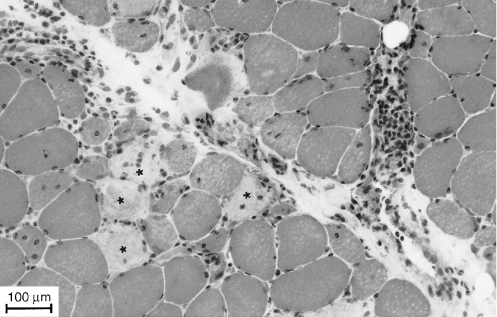
The diagnosis is confirmed by the muscle biopsy findings. Characteristic features include perifascicular atrophy, focal myofibrillar loss and areas of infarction. Vascular changes include capillary necrosis, undulating tubules in endothelial cells, arteriolar thrombosis and lytic membrane-attack complex deposition in vessel walls. Inflammatory infiltrates are typically found in septa and around blood vessels (Figure 67.2) and consist of B-lymphocytes, helper T-lymphocytes and plasma cells.
Figure 67.2 Dermatomyositis. Muscle biopsy (H&E, ×115). Note the perivascular inflammatory infiltrate and necrotic fibres (*).
Pathogenesis
On the basis of the characteristic immunopathological muscle biopsy findings noted above, it is believed that DM is caused by humoral immune mechanisms which lead to destruction of capillaries and occlusion of arterioles. Muscle fibre damage is thus secondary to ischaemia. This contrasts with PM in which cell-mediated immune mechanisms lead to muscle fibre destruction.
Treatment
Knowledge of the natural history of DM is limited and there have been no completely satisfactory controlled trials of drug therapy. Prednisolone is the mainstay of treatment and a reasonable starting dose is 1 mg kg−1 body weight per day. In severe cases, this may be preceded by intravenous methylprednisolone 500 mg daily for 5 days, although the evidence for additional benefit is lacking. Azathioprine (2.5 mg kg−1 body weight per day) or methotrexate (10–20 mg weekly) are widely used in combination with prednisolone, as long-term steroid-sparing agents. The steroid dose is maintained for 1–3 months (at least until the CK has returned to normal) and then lowered slowly at a rate determined by the clinical response and to a lesser extent by changes in the serum CK. A rise in the CK may precede exacerbation of weakness. As the disorder settles, the prednisolone can be changed to an alternate day regime, which may reduce steroid side effects. If the patient does not respond to or cannot tolerate the above regime, then there are several other options. Alternative immunosuppressant drugs that are used (but without trial data) include mycophenolate mofetil and cyclophosphamide. Plasma exchange and leucapheresis appear to be ineffective. Intravenous immunoglobulin is almost certainly effective,9 but there are insufficient data to recommend it over prednisolone as a mainstay of treatment, although it is fairly widely used in severe cases at first presentation together with high doses of steroids.
The cutaneous lesions respond to systemic steroid treatment. In the absence of significant muscle involvement, topical steroids may have a role. The skin is photosensitive and light-exposed areas should be treated with an ultraviolet blocking cream. Malnutrition (e.g. due to associated dysphagia) must be avoided to prevent muscle catabolism. Physical activity should be encouraged.
The prognosis with respect to muscle function and life expectancy is good if treatment is started early and there is no associated malignancy.
Polymyositis (PM)
Clinical Features
Polymyositis is a disappearing disease as many cases previously diagnosed as PM have been reclassified as having IBM or myositis associated with connective tissue disease.10 As in DM, the weakness is predominantly proximal but the onset is often insidious and the rate of progression slower. Muscle pain and tenderness are very uncommon. Elderly patients often give a history of deterioration of gait over 1–2 years, which is not infrequently attributed to arthritic hips.
Diagnosis and Pathological Features
The serum CK is usually elevated but may be normal in very slowly progressive or late stages of the disease. The ESR is unhelpful. Electromyographic changes are the same as those described for DM.
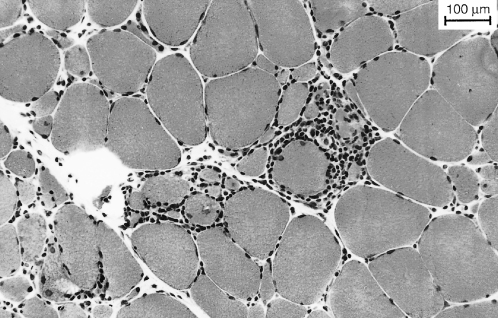
Muscle biopsy shows scattered necrotic and regenerating fibres. The vascular changes noted in DM are absent. Inflammatory infiltrates (Figure 67.3) tend to be within fascicles (endomysial) and are composed of cytotoxic T-lymphocytes and macrophages with fewer B-lymphocytes. A characteristic finding in PM, and also in IBM, is partial invasion of muscle fibres. Cytotoxic T-lymphocytes penetrate the basal lamina but not the muscle fibre membrane and appear to compress the fibres without causing necrosis. These fibres, and also non-invaded fibre, express class I major histocompatibility complex (MHC) protein products, which are not expressed in normal muscle.
Pathogenesis
In PM, it is presumed that cytotoxic T-lymphocytes recognize an antigen, bound to class I MHC products, on the muscle fibre surface. Muscle fibre function is compromised by the invading lymphocytes, which are also associated with lymphokine release. The nature and origin of the antigen are unknown.
Treatment
Despite the different pathogenetic mechanisms postulated, the immunosuppressive drug management of PM is the same as described above for DM. If there is extensive weakness and wasting at presentation, the prognosis for significant improvement is poor, although disease progress may be arrested.
Inclusion Body Myositis (IBM)
Clinical Features
IBM is rare before the fifth or sixth decade of life and shows a strong male predominance.11 Its incidence is uncertain and many cases have previously been labelled as (steroid-resistant) PM. Rare familial cases have been described.12
The characteristic pattern of muscle involvement, which is often asymmetric, involves wasting and weakness of quadriceps and distal weakness affecting finger flexion and ankle dorsiflexion. Symptomatic presentation is with falls due to the knees giving way and weakness of grip.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


