Chromosomal Translocations in Multiple Myeloma
Mutated Genes in Multiple Myeloma
Prognostic Implications of Genetic Lesions
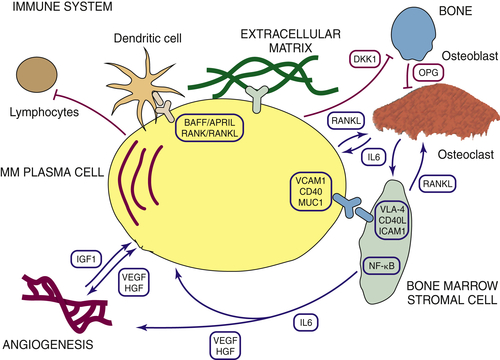
The Microenvironment in Multiple Myeloma
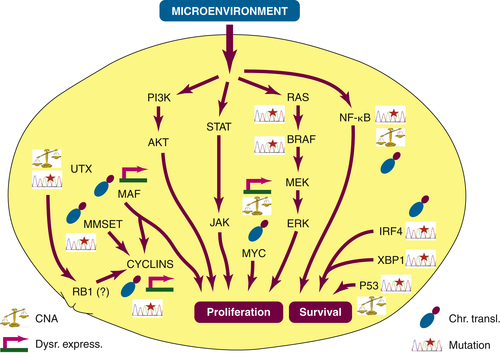
Deregulated Pathways in Myeloma and the Opening of Novel Therapeutic Opportunities
MYC
MAF
Cyclins
Chromatin Remodeling Genes
MMSET
UTX
RB1, P18, and Other Members of the RB1 Pathway
NFκB
TP53 Deletion and Mutations
The Proteasome Achilles’ Heel
The Molecular Basis of the Evolution from MGUS to MM
Final Remarks
Acknowledgments
1. Clinical factors predictive of outcome with bortezomib in patients with relapsed, refractory multiple myeloma . Blood . 2005 ; 106 : 2977–2981 .
2. Novel biological therapies for the treatment of multiple myeloma . Best Pract Res Clin Haematol . 2005 ; 18 : 619–634 .
3. Treatment of multiple myeloma . Blood . 2004 ; 103 : 20–32 .
4. Multiple myeloma . N Engl J Med . 2011 ; 364 : 1046–1060 .
5. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study . Blood . 2009 ; 113 : 5412–5417 .
6. A monoclonal gammopathy precedes multiple myeloma in most patients . Blood . 2009 ; 113 : 5418–5422 .
7. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management . Leukemia . 2010 ; 24 : 1121–1127 .
8. Pathogenesis of monoclonal gammopathy of undetermined significance and progression to multiple myeloma . Semin Hematol . 2011 ; 48 : 4–12 .
9. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma . Blood . 2005 ; 106 : 296–303 .
10. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients . Cancer Cell . 2006 ; 9 : 313–325 .
11. The molecular classification of multiple myeloma . Blood . 2006 ; 108 : 2020–2028 .
12. Multiple myeloma: evolving genetic events and host interactions . Nat Rev Cancer . 2002 ; 2 : 175–187 .
13. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases . Leukemia . 1998 ; 12 : 960–969 .
14. The genetic architecture of multiple myeloma . Nat Rev Cancer . 2012 ; 12 : 335–348 .
15. Acute lymphoblastic leukemia . N Engl J Med . 2004 ; 350 : 1535–1548 .
16. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS . Blood . 2005 ; 106 : 2156–2161 .
17. Ploidy status rarely changes in myeloma patients at disease progression . Leuk Res . 2006 ; 30 : 266–271 .
18. Chromosome translocations in multiple myeloma . Oncogene . 2001 ; 20 : 5611–5622 .
19. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma . Proc Natl Acad Sci U S A . 1996 ; 93 : 13931–13936 .
20. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma . Leukemia . 2003 ; 17 : 427–436 .
21. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma . Blood . 2003 ; 102 : 2562–2567 .
22. Multiple myeloma primary cells show a highly rearranged unbalanced genome with amplifications and homozygous deletions irrespective of the presence of immunoglobulin-related chromosome translocations . Haematologica . 2007 ; 92 : 795–802 .
23. Identification of overexpressed genes in frequently gained/amplified chromosome regions in multiple myeloma . Haematologica . 2006 ; 91 : 184–191 .
24. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma . Blood . 2006 ; 108 : 1733–1743 .
25. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome . Clin Cancer Res . 2010 ; 16 : 1856–1864 .
26. Gene rearrangement and overexpression of PRAD1 in lymphoid malignancy with t(11;14)(q13;q32) translocation . Oncogene . 1992 ; 7 : 1401–1406 .
27. Genomic abnormalities in monoclonal gammopathy of undetermined significance . Blood . 2002 ; 100 : 1417–1424 .
28. 14q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Intergroupe Francophone du Myelome . Cancer Res . 1999 ; 59 : 4546–4550 .
29. Myeloma and the t(11;14)(q13;q32): evidence for a biologically defined unique subset of patients . Blood . 2002 ; 99 : 3735–3741 .
30. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation . Blood . 2002 ; 99 : 2185–2191 .
31. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma . Blood . 2001 ; 98 : 217–223 .
32. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma? Leuk Lymphoma . 2006 ; 47 : 2289–2300 .
33. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients . Blood . 2005 ; 105 : 4060–4069 .
34. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma . Blood . 2001 ; 97 : 729–736 .
35. Analysis of FGFR3 gene mutations in multiple myeloma patients with t(4;14) . Br J Haematol . 2001 ; 114 : 362–364 .
36. C-MAF oncogene dysregulation in multiple myeloma: frequency and biological relevance . Leuk Lymphoma . 2003 ; 44 : 1761–1766 .
37. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma . Blood . 1998 ; 91 : 4457–4463 .
38. Ectopic expression of MAFB gene in human myeloma cells carrying (14;20)(q32;q11) chromosomal translocations . Jpn J Cancer Res . 2001 ; 92 : 638–644 .
39. Initial genome sequencing and analysis of multiple myeloma . Nature . 2011 ; 471 : 467–472 .
40. Exploring the genomes of cancer cells: progress and promise . Science . 2011 ; 331 : 1553–1558 .
41. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses . Science . 2008 ; 321 : 1801–1806 .
42. Frequent engagement of the classical and alternative NFkappaB pathways by diverse genetic abnormalities in multiple myeloma . Cancer Cell . 2007 ; 12 : 115–130 .
43. Promiscuous mutations activate the noncanonical NFkappaB pathway in multiple myeloma . Cancer Cell . 2007 ; 12 : 131–144 .
44. IRF4 addiction in multiple myeloma . Nature . 2008 ; 454 : 226–231 .
45. Hypodiploidy and 22q11 rearrangements at diagnosis are associated with poor prognosis in patients with multiple myeloma . Br J Haematol . 1997 ; 98 : 418–425 .
46. Both hypodiploidy and deletion of chromosome 13 independently confer poor prognosis in multiple myeloma . Br J Haematol . 2002 ; 118 : 1041–1047 .
47. Hypodiploidy is a major prognostic factor in multiple myeloma . Blood . 2001 ; 98 : 2229–2238 .
48. Upregulation of translational machinery and distinct genetic subgroups characterise hyperdiploidy in multiple myeloma . Br J Haematol . 2007 ; 136 : 565–573 .
49. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling . Cancer Res . 2007 ; 67 : 2982–2989 .
50. Prognostic factors for hyperdiploid-myeloma: effects of chromosome 13 deletions and IgH translocations . Leukemia . 2006 ; 20 : 807–813 .
51. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma . Leukemia . 2006 ; 20 : 2034–2040 .
52. Significant increase of CKS1B amplification from monoclonal gammopathy of undetermined significance to multiple myeloma and plasma cell leukaemia as demonstrated by interphase fluorescence in situ hybridisation . Br J Haematol . 2006 ; 134 : 613–615 .
53. Abnormalities of chromosome 1p/q are highly associated with chromosome 13/13q deletions and are an adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma . Br J Haematol . 2007 ; 136 : 615–623 .
54. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1 . Blood . 2007 ; 109 : 2276–2284 .
55. High incidence of chromosome 13 deletion in multiple myeloma detected by multiprobe interphase FISH . Blood . 2000 ; 96 : 1505–1511 .
56. Clinical and biologic implications of recurrent genomic aberrations in myeloma . Blood . 2003 ; 101 : 4569–4575 .
57. Deletion of 13q14 remains an independent adverse prognostic variable in multiple myeloma despite its frequent detection by interphase fluorescence in situ hybridization . Blood . 2000 ; 95 : 1925–1930 .
58. Deletions of chromosome 13q in monoclonal gammopathy of undetermined significance . Leukemia . 2000 ; 14 : 1975–1979 .
59. The t(4;14)(p16.3;q32) is strongly associated with chromosome 13 abnormalities in both multiple myeloma and monoclonal gammopathy of undetermined significance . Blood . 2001 ; 98 : 1271–1272 .
60. Results of high-dose therapy for 1000 patients with multiple myeloma: durable complete remissions and superior survival in the absence of chromosome 13 abnormalities . Blood . 2000 ; 95 : 4008–4010 .
61. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities . Blood . 1995 ; 86 : 4250–4256 .
62. Chromosome 13 abnormalities identified by FISH analysis and serum beta2-microglobulin produce a powerful myeloma staging system for patients receiving high-dose therapy . Blood . 2001 ; 97 : 1566–1571 .
63. Biological and prognostic significance of interphase fluorescence in situ hybridization detection of chromosome 13 abnormalities (delta13) in multiple myeloma: an Eastern Cooperative Oncology Group study . Cancer Res . 2002 ; 62 : 715–720 .
64. Continuous absence of metaphase-defined cytogenetic abnormalities, especially of chromosome 13 and hypodiploidy, ensures long-term survival in multiple myeloma treated with Total Therapy I: interpretation in the context of global gene expression . Blood . 2003 ; 101 : 3849–3856 .
65. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib . Blood . 2007 ; 109 : 3177–3188 .
66. The microenvironment in mature B-cell malignancies: a target for new treatment strategies . Blood . 2009 ; 114 : 3367–3375 .
67. Bone marrow microenvironment and the identification of new targets for myeloma therapy . Leukemia . 2009 ; 23 : 10–24 .
68. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets . Nat Rev Cancer . 2007 ; 7 : 585–598 .
69. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma . Blood . 1999 ; 93 : 3064–3073 .
70. Bone marrow angiogenesis in multiple myeloma . Leukemia . 2006 ; 20 : 193–199 .
71. Targeting multiple-myeloma-induced immune dysfunction to improve immunotherapy outcomes . Clin Dev Immunol . 2012 ; 2012 : 196063 .
72. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target . Cancer Cell . 2009 ; 16 : 309–323 .
73. Heterogeneous pattern of chromosomal breakpoints involving the MYC locus in multiple myeloma . Genes Chromosomes Cancer . 2003 ; 37 : 261–269 .
74. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma . Proc Natl Acad Sci U S A . 2000 ; 97 : 228–233 .
75. Complex translocation disrupts c-myc regulation in a human plasma cell myeloma . Mol Cell Biol . 1988 ; 8 : 124–129 .
76. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors . Blood . 2001 ; 98 : 3082–3086 .
77. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma . Leukemia . 2011 ; 25 : 1026–1035 .
78. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies . Cancer Cell . 2008 ; 13 : 167–180 .
79. Transcription factors as targets for cancer therapy . Nat Rev Cancer . 2002 ; 2 : 740–749 .
80. BET bromodomain inhibition as a therapeutic strategy to target c-Myc . Cell . 2011 ; 146 : 904–917 .
81. ARK5 is transcriptionally regulated by the Large-MAF family and mediates IGF-1-induced cell invasion in multiple myeloma: ARK5 as a new molecular determinant of malignant multiple myeloma . Oncogene . 2005 ; 24 : 6936–6944 .
82. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma . Cancer Cell . 2004 ; 5 : 191–199 .
83. Molecular characterization of human multiple myeloma cell lines by integrative genomics: insights into the biology of the disease . Genes Chromosomes Cancer . 2007 ; 46 : 226–238 .
84. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression . Blood . 2011 ; 117 : 2396–2404 .
85. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma . N Engl J Med . 2003 ; 349 : 2483–2494 .
86. Osteopontin dysregulation and lytic bone lesions in multiple myeloma . Hematol Oncol . 2007 ; 25 : 16–20 .
87. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines . Blood . 1996 ; 88 : 674–681 .
88. Messenger RNA levels of five genes located at chromosome 11q13 in B-cell tumors with chromosome translocation t(11;14)(q13;q32) . Cancer Res . 1994 ; 54 : 377–379 .
89. Overexpression of the PRAD1 oncogene in a patient with multiple myeloma and t(11;14)(q13;q32) . Acta Haematol . 1995 ; 94 : 199–203 .
90. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3 . Blood . 2011 ; 118 : 3512–3524 .
91. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma . Science . 2010 ; 330 : 228–231 .
92. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers . Nat Rev Cancer . 2010 ; 10 : 457–469 .
93. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3 . Nat Genet . 1997 ; 16 : 260–264 .
94. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts . Blood . 1998 ; 92 : 3025–3034 .
95. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor . Blood . 2008 ; 111 : 3145–3154 .
96. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells . Blood . 2011 ; 117 : 211–220 .
97. Transcription repression activity is associated with the type I isoform of the MMSET gene involved in t(4;14) in multiple myeloma . Br J Haematol . 2005 ; 131 : 214–218 .
98. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity . Mol Cell Biol . 2008 ; 28 : 2023–2034 .
99. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma . Blood . 2011 ; 117 : 553–562 .
100. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites . Nature . 2011 ; 470 : 124–128 .
101. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) . Blood . 2002 ; 99 : 3857–3860 .
102. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines . Genomics . 2001 ; 74 : 79–88 .
103. High-resolution genomic profiles of human lung cancer . Proc Natl Acad Sci U S A . 2005 ; 102 : 9625–9630 .
104. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia . Blood . 2001 ; 98 : 1264–1267 .
105. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer . Nat Genet . 2009 ; 41 : 521–523 .
106. Drosophila UTX is a histone H3 Lys27 demethylase that colocalizes with the elongating form of RNA polymerase II . Mol Cell Biol . 2008 ; 28 : 1041–1046 .
107. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling . Cancer Cell . 2011 ; 19 : 86–100 .
108. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer . Science . 2008 ; 322 : 1695–1699 .
109. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells . Blood . 2002 ; 99 : 1745–1757 .
110. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas . Proc Natl Acad Sci U S A . 2010 ; 107 : 20980–20985 .
111. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation . Blood . 2011 ; 117 : 2451–2459 .
112. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype . Oncogene . 2005 ; 24 : 6269–6280 .
113. The histone demethylase UTX enables RB-dependent cell fate control . Genes Dev . 2010 ; 24 : 327–332 .
114. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development . Nature . 2007 ; 449 : 731–734 .
115. Expression of HOXA genes in patients with multiple myeloma . Leuk Lymphoma . 2004 ; 45 : 1215–1217 .
116. Gene abnormalities in multiple myeloma: the relevance of TP53, MDM2, and CDKN2A . Haematologica . 2003 ; 88 : 529–537 .
117. Frequent hypermethylation of p16 and p15 genes in multiple myeloma . Blood . 1997 ; 89 : 2500–2506 .
118. Tumor suppressor p16 methylation in multiple myeloma: biological and clinical implications . Blood . 2007 ; 109 : 1228–1232 .
119. Methylation and expression of the p16INK4A tumor suppressor gene in multiple myeloma . Blood . 2007 ; 109 : 1337–1338 .
120. p16 gene methylation lacks correlation with angiogenesis and prognosis in multiple myeloma . Cancer Lett . 2005 ; 222 : 247–254 .
121. Alterations of the cyclin D1/pRb/p16(INK4A) pathway in multiple myeloma . Leukemia . 2002 ; 16 : 1844–1851 .
122. Paradoxical expression of INK4c in proliferative multiple myeloma tumors: bi-allelic deletion vs increased expression . Cell Div . 2006 ; 1 : 23 .
123. Analysis of the p16INK4A, p15INK4B and p18INK4C genes in multiple myeloma . Br J Haematol . 1997 ; 96 : 98–102 .
124. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival . Clin Cancer Res . 2011 ; 17 : 7776–7784 .
125. Classical and/or alternative NFkappaB pathway activation in multiple myeloma . Blood . 2010 ; 115 : 3541–3552 .
126. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy . Blood . 1993 ; 81 : 128–135 .
127. Rare occurrence of P53 gene mutations in multiple myeloma . Br J Haematol . 1992 ; 81 : 440–443 .
128. p53 and RAS gene mutations in multiple myeloma . Oncogene . 1992 ; 7 : 2539–2543 .
129. Inactivation of tumor suppressor genes, p53 and Rb1, in plasma cell dyscrasias . Leukemia . 1994 ; 8 : 758–767 .
130. Mutations of the p53 gene in human myeloma cell lines . Oncogene . 1992 ; 7 : 1015–1018 .
131. Clinical significance of TP53 mutation in myeloma . Leukemia . 2007 ; 21 : 582–584 .
132. P53 deletion is not a frequent event in multiple myeloma . Br J Haematol . 1999 ; 106 : 717–719 .
133. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome . Blood . 2007 ; 109 : 3489–3495 .
134. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation . Blood . 2005 ; 105 : 358–360 .
135. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy . Blood . 2005 ; 106 : 2837–2840 .
136. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis . Blood . 2003 ; 102 : 4504–4511 .
137. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells . Cancer Res . 2001 ; 61 : 3071–3076 .
138. Both IGH translocations and chromosome 13q deletions are early events in monoclonal gammopathy of undetermined significance and do not evolve during transition to multiple myeloma . Leukemia . 2004 ; 18 : 1879–1882 .
139. Evaluation of multiple models to distinguish closely related forms of disease using DNA microarray data: an application to multiple myeloma . Stat Appl Genet Mol Biol . 2004 ; 3 (10) .
140. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis . Blood . 2007 ; 109 : 1692–1700 .
141. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells . Blood . 2007 ; 110 : 2586–2592 .
142. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance . Blood . 2005 ; 106 : 812–817 .
143. Oncogenes in multiple myeloma: point mutation of N-ras . Oncogene . 1990 ; 5 : 1659–1663 .
144. Ras oncogene mutation in multiple myeloma . J Exp Med . 1989 ; 170 : 1715–1725 .
145. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis . Hum Mutat . 2001 ; 18 : 212–224 .
146. Activating mutations of N- and K-ras in multiple myeloma show different clinical associations: analysis of the Eastern Cooperative Oncology Group Phase III Trial . Blood . 1996 ; 88 : 2699–2706 .
147. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias . Blood . 1993 ; 81 : 2708–2713 .
148. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors . Blood . 2005 ; 105 : 317–323 .
149. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression . Blood . 2003 ; 101 : 1520–1529 .
150. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by reverse transcription-polymerase chain reaction analysis of IGH-MMSET fusion transcripts . Cancer Res . 2000 ; 60 : 4058–4061 .
151. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides . Blood . 2012 ; 120 : 1060–1066 .
152. Clonal competition with alternating dominance in multiple myeloma . Blood . 2012 ; 120 : 1067–1076 .
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


