Abstract
Multiple endocrine neoplasia type 1 is an autosomal dominantly inherited syndrome. MEN1 is characterized by the occurrence of tumors of the parathyroid glands, the pancreatic islets, the anterior pituitary gland, and the adrenal glands, as well as neuroendocrine tumors in the thymus, lungs, and stomach, often at a young age. Nonendocrine manifestations of MEN1 include angiofibromas, collagenomas, lipomas, leiomyomas, and meningiomas. The prevalence of MEN1 is two to three per 100,000, and is equal among males and females. MEN1 and multiple endocrine neoplasia type 2 (MEN2) are two distinct syndromes. In MEN2, patients frequently develop medullary thyroid carcinoma and adrenal medullary tumors (pheochromocytoma).
Keywords
multiple endocrine neoplasia type 1, MEN1, mutation analysis, periodical monitoring, menin, genetic testing
Introduction
Background, Prevalence
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominantly inherited syndrome. MEN1 is characterized by the occurrence of tumors of the parathyroid glands, the pancreatic islets, the anterior pituitary gland, and the adrenal glands, as well as neuroendocrine tumors (NETs) in the thymus, lungs, and stomach, often at a young age. Nonendocrine manifestations of MEN1 include angiofibromas, collagenomas, lipomas, leiomyomas, and meningiomas ( Table 24.1 ).
| A. Endocrine lesions | |
| Parathyroid adenomas | 75–95% |
| Pancreatic islet cell tumors | 55% |
| Gastrinomas | 45% |
| Insulinomas | 10% |
| No clear clinical picture (including PP-, SS-producing tumors) | 10–80% |
| Other (VIP, GHRH, etc.) | 2% |
| Pituitary adenomas | 47% |
| Prolactinomas | 30% |
| Nonfunctioning (i.e., not producing hormone) | 10% |
| ACTH producing | 1% |
| GH producing | 3–6% |
| Adrenal cortical adenomas | 20% |
| NETs | 18% |
| Thymus | 1% |
| Lungs | 12% |
| Stomach | 5% |
| B. Nonendocrine lesions | |
| Skin lesions | 80% |
| Angiofibromas | 75% |
| Collagenomas | 5% |
| Lipomas | 30% |
| Leiomyomas | 5% |
| Meningiomas | 25% |
The prevalence of MEN1 is 2–3 per 100,000, and is equal among males and females.
MEN1 and multiple endocrine neoplasia type 2 (MEN2) are two distinct syndromes. In MEN2, patients frequently develop medullary thyroid carcinoma and adrenal medullary tumors (pheochromocytoma).
Natural History
From family studies in the past, it appeared that if no treatment is given, life expectancy is considerably shortened.
In MEN1, the median life expectancy for patients with a malignant islet cell tumor is 46 years, for peptic ulcer disease 56 years, for malignant carcinoid 53 years, for hypercalcemia/uremia 42 years, and the overall median age at death for MEN1 is 47 years.
MEN1 is caused by germline mutations of the MEN1 gene. Since the discovery of the gene in 1997, DNA diagnosis has become available. Carriers of a MEN1 gene germline mutation can be monitored periodically by targeted clinical examination to identify MEN1-associated lesions at a presymptomatic stage.
In this review, the recent developments concerning the etiology of MEN1 as well as the current diagnostic and therapeutic options are presented. Furthermore, guidelines for MEN1 gene mutation analysis and periodic clinical monitoring are provided.
Clinical Presentation, Diagnosis, and Treatment
According to the MEN consensus published in 2001 and updated in 2012, a practical definition of a MEN1 patient is a patient with at least two of the three main MEN1-related endocrine tumors (i.e., parathyroid adenomas, enteropancreatic endocrine tumors, and a pituitary adenoma). Familial MEN1 is similarly defined as at least one MEN1 case plus at least one first degree relative with one of those main MEN1 tumors. Alternatively, because parathyroid and pituitary adenomas occur relatively frequently in the general population, a MEN1 patient can be defined more precisely as a patient with three or more of the five major MEN1-associated lesions (i.e., besides the three tumor types mentioned, adrenal gland tumors, and NETs in the thymus, lungs, and stomach). A MEN1-suspected patient is defined as having at least two major MEN1-associated lesions, multiple lesions within one MEN1-related organ, and/or a MEN1-associated lesion at a young age (<35 years). Such a patient may be considered for DNA analysis (see under MEN1 mutation analysis).
Below, for each tumor type the clinical presentation and the diagnostic and therapeutic options are listed. In Figure 24.1 a–c, flow charts are shown for diagnosis and therapy of MEN1-associated tumors of the parathyroid glands, pancreatic islets, and pituitary adenoma.
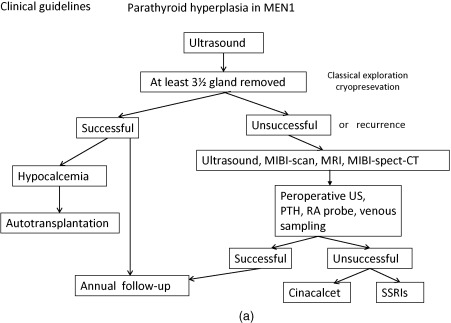
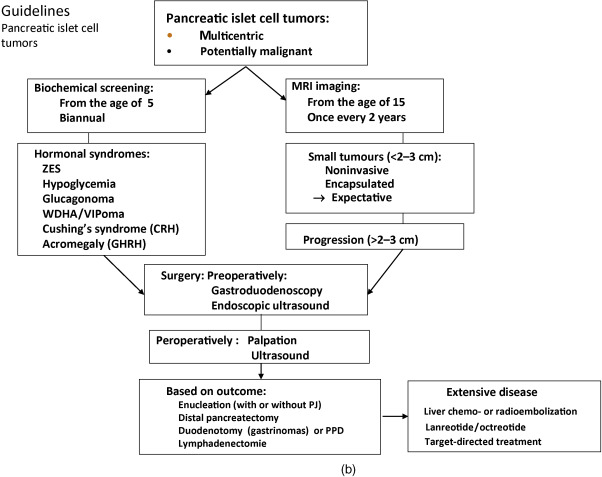
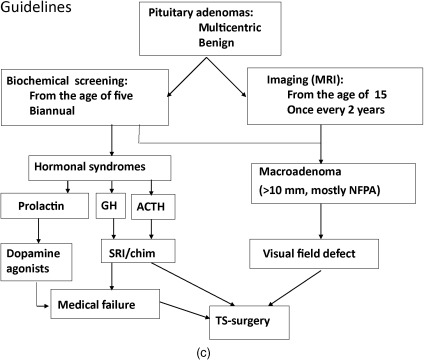
It may explain the variable expression and the significance of general guidelines.
Parathyroid Adenoma
Parathyroid adenomas are often the first manifestation of MEN1. About 75–95% of MEN1 patients develop parathyroid adenomas. Usually, parathyroid adenomas in MEN1 are multiple and benign.
Phenocopies
Other forms of familial hyperparathyroidism (FHPT) or phenocopies of MEN1 are found in familial isolated hyperparathyroidism (FIHP). In some FIHP families, a CASR mutation may be identified. The FHPT jaw tumor syndrome (HPT-JT, HRPT2) is caused by inactivating germline mutations in the parafibromin gene on chromosome 1 (1q31). FHPT in MEN2A is caused by activating germline mutations in the RET proto-oncogene on chromosome 10 (10q11.2), and in MEN4, it is caused by inactivating germline mutations in the CDKN1B gene encoding the p27 protein. MEN1 genotyping appears worthwhile in FIHP families, as the finding of MEN1 gene mutation(s) in this gene predicts possible involvement of other organs.
The increased production of parathyroid hormone causes hypercalcemia. Symptoms and signs of hypercalcemia include fatigue, depression, constipation, nausea, symptoms caused by nephrolithiasis or nephrocalcinosis, bone pain, myalgia, and arthralgia as well as hypertension (see Fig. 24.1 a).
Diagnosis
Laboratory investigation consists of measurement of (ionized) calcium, chloride, phosphate, and parathyroid hormone in blood. To distinguish primary hyperparathyroidism from hypocalciuric hypercalcemia, in addition to this, the 24-h calcium excretion in the urine is measured. Bone densitometry can be used to detect bone mass reduction.
Preoperative localization is useful, because adenomas may be situated in or behind the thyroid lobes.
Parathyroid adenomas can be effectively localized by a nuclear scan with Tc-99m sestamibi, which is retained selectively by parathyroid adenoma. To confirm the location, ultrasound (US) and/or computed tomography (CT) can be performed (see Fig. 24.1 a). Preoperative imaging in PHP in MEN1 is strictly not indicated as it will not alter the operative strategy (i.e., conventional neck exploration). One might consider Tc-99m sestamibi scintigraphy to exclude ectopic adenomas, but this is by no means standard practice.
Subtotal parathyroidectomy with bilateral transcervical thymectomy is the procedure of choice for MEN1-related hyperparathyroidism. Total parathyroidectomy has lowest risk of persistent and recurrent pHPT. However, total parathyroidectomy had the highest risk of permanent hypoparathyroidism; a less than subtotal parathyroidectomy has an unacceptable high rate of recurrent and persistent pHPT. Performing intraoperative confirmation by histologic examination and rapid parathyroid hormone assay with autologous graft of parathyroid tissue have the lowest recurrence rate.
In most cases, this surgical approach was able to restore normal calcium/parathyroid hormone levels improved bone mineral density and ultimately lead to discontinuation of calcium and calcitriol supplementation. Long-term follow-up is recommended.
Tumors of the Endocrine Duodenum and Pancreatic Islets
Definition Neuroendocrine Tumors
In the past, the classical term “carcinoid” was well established in medical terminology; however, at present, it is not adequate to cover the entire spectrum of neuroendocrine neoplasms.
In 2000, the term NETs was suggested by the World Health Organization (WHO) in the new classification of tumors that differ in their morphological and functional features.
Divergence in gene-expression patterns in the development of tumors from the gastroenteropancreatic (GEP) system (divided in NETs of jejunum and ileum and pancreaticoduodenal endocrine tumors (PETs)) was identified, when they were examined at a molecular level. On the basis of gene expression profiles, neuroendocrine lesions of jejunum and ileum and PETs need to be considered as two distinct entities within the group of NETs.
PETs develop in about 55% of MEN1 patients. Multicentric microadenomas are present in 90% of MEN1 patients. In 44% of sporadically occurring PETs, a somatic inactivating mutation of the MEN1 gene was identified.
Symptomatology
Hormonal syndromes often occur late and indicate metastases in 50% of patients with this stage of functioning PETs. Prospective screening with biochemical markers and endoscopic ultrasound (EUS) is therefore recommended. The use of biochemical tumor markers for diagnosing PETs has been recently debated. Prospective endoscopic ultrasonic evaluation reveals that the frequency of nonfunctioning PETs is higher (55%) than previously thought (34%).
Absence of Symptoms
It was demonstrated that high penetrance of NF-PETs occurs in 15–20-year-old MEN1 patients. The high percentage of the patients presenting consensus criteria for surgery for NF-PET alone or NF-PET/insulinoma suggests a potential benefit for the periodic surveillance of these tumors in this age group.
Nonfunctioning pancreatic NETs may occur in asymptomatic children with MEN1 mutations, and screening for such enteropancreatic tumors in MEN1 children should be considered earlier than the age of 20 years, as is currently recommended by the international guidelines.
Small asymptomatic neuroendocrine pancreatic tumors in MEN1 usually seem to grow slowly. Annual tumor incidence rate is low. However, faster growing tumors and patients with rapidly progressive disease can be observed. EUS is a sensitive method to detect these tumors.
Diagnosis
Laboratory investigation includes determination of fasting plasma levels of glucose, insulin, C-peptide, glucagon, gastrin, pancreatic polypeptide, and chromogranin A.
Pancreatic islet cell tumors can be visualized by MRI, somatostatin receptor scintigraphy, Ga-68-DOTATATE scintigraphy CT, and F-DOPA positron emission tomography (PET) (see Fig. 24.1 b).
EUS is useful for early detection of PETs and will allow early surgery before metastases have developed.
EUS is a more sensitive technique than CT or transabdominal US for the detection and localization of potentially malignant lesions in patients with MEN1.
Selective arterial secretagogue injection test (SASI test) may be useful for localizing functioning pNETs. In case of a functioning pNET, the tumor should first be accurately located using the SASI test before an appropriate surgical method is selected.
Specific Syndromes
Zollinger–Ellison Syndrome (ZES)
In MEN1, gastrinomas are most often located submucosally in the antrum and duodenum.
The elevated levels of ectopic gastrin cause excessive gastric acid production. If untreated, this can lead to the ZES: ulcerations of the digestive tract, diarrhoea, and mucosal hypertrophy. Before treatment with proton pump inhibitors became available, the ZES was a frequent cause of death of MEN1 patients. Gastrinomas are still an important threat to MEN1 patients, because they are often multicentric and are able to metastasize to the lymph nodes and the liver.
The diagnosis is delayed by the widespread use of proton pump inhibitors.
MEN1 patients frequently develop ZES. About 25% of all ZES patients have MEN1. Esophageal reflux symptoms are common resulting in strictures and Barrett’s esophagus (BE). The frequency of severe peptic esophageal disease, including the premalignant condition BE, was higher in MEN1 patients with gastrinomas than in patients with sporadic gastrinomas. This higher incidence of severe esophageal disease in MEN1/ZES was due to delay of diagnosis, more frequent and severe esophageal symptoms, more frequent hiatal hernias, more common pyloric scarring, higher basal acid output, and underdiagnosed hyperparathyroidism. There may be a relationship between hyperparathyroidism and the development of ZES and gastric NETs from enterochromaffin-like (ECL) cells (see Fig. 24.2 ).
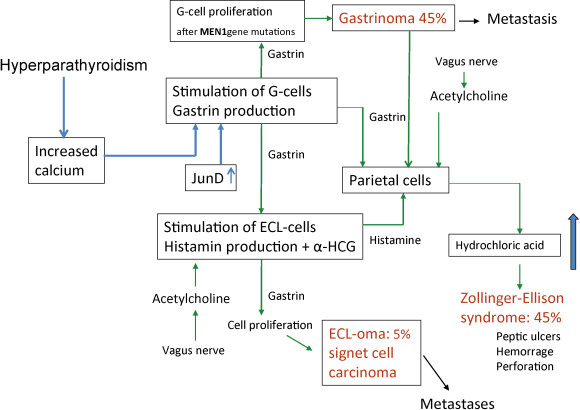
Recurrence may occur by ectopic location. These findings suggest the importance of checking for the presence of ectopic gastrinomas in the biliary tree in MEN1 patients undergoing ZES surgery.
Surgery
While small lesions may be treated conservatively, most larger lesions or those refractory to medical treatment should be considered for surgery. As submucosal duodenal lesions are often present, the standard approach involves a pylorus-preserving pancreaticoduodenectomy. In cases where less aggressive surgery is warranted, duodenal exploration should always be part of the procedure.
A suggested procedure includes distal 80% subtotal pancreatic resection together with enucleation of tumors in the head of the pancreas, and in cases with ZES, excision of duodenal gastrinomas together with clearance of regional lymph node metastases. This strategy, with early and aggressive surgery before metastases have developed, is believed to reduce the risks for tumor recurrence and malignant progression. Given the shortage of evidence from controlled clinical trials, the preferable surgical strategy for patients with gastrinoma is unclear. The treatment is controversial. The strategy should be adapted to the characteristics of the individual patient, for example, age, medical history, comorbidity, and preferably discussed within an experienced multidisciplinary team. Improved long-term survival is obtained by curative surgery for patients with MEN1-associated GEPNET. The current surgical indications are expanding even in patients with hepatic metastases because of the improved surgical outcome.
Other Functioning Pancreatic Islet Cell Tumors
Insulinomas occur in about 10% of all MEN1 patients. They may present with symptoms of hypoglycemia, such as confusion or abnormal behavior, due to central nervous system dysfunction at times of exersise or fasting.
Pasireotide (SOM230) demonstrates antisecretory, antiproliferative, and proapoptotic activity in a MEN1 knockout mouse model of insulinoma. Further studies of the effects of SOM230 in pNET patients with MEN1 mutations are needed to establish the role of SOM230 in patients.
VIP, Somatstatin, Glucagonoma, and WDHA
Infrequently occurring syndromes are glucagonoma (1.6%), VIPoma (0.98%), and somatostatinoma (0.65%). Glucagonomas can cause skin lesions, whereas tumors producing vasoactive intestinal peptide (VIP), VIPomas, can cause the Verner–Morrison syndrome; also mentioned is the watery-diarrhea-hypokalemia-achlorhydria (WDHA) syndrome. Surgical excision of insulinomas, glucagonomas, and VIPomas is usually curative. Tumors producing pancreatic polypeptide are common (>80% of pNETs), but only rarely cause symptoms and therefore do normally not require treatment.
Growth Hormone-Releasing Hormone (GHRH)-Producing Tumors
Acromegaly may be caused by growth hormone-releasing hormone (GHRH) produced by pancreatic islet cell tumors. GHRH may cause acromegaly indirectly through stimulation of growth hormone (GH) production by the pituitary gland. In more than 50% of MEN1 patients with acromegaly, a GHRH-producing pancreatic tumor is the cause of the disease.
In MEN1 patients, acromegaly can also be caused by adenomas in the pituitary gland, primarily producing GH, and NETs in the thymus gland producing GHRH.
Pituitary Adenomas
Evidence of MEN1 is found in approximately 2.7% of all patients with pituitary adenomas. In addition, somatic mutations in the MEN1 gene do not play a prominent role in the pathogenesis of sporadic forms of pituitary adenoma (3.7%).
Pituitary tumors in MEN1 are larger in size and more aggressive than sporadic tumors. MEN1 must be considered in all children with tumors of the pituitary gland.
The most frequently occurring pituitary tumors in MEN1 are prolactinomas. Prolactinomas occur in approximately 30% of patients with MEN1 and in this setting, they may be more aggressive than their sporadic counterparts.
A MEN1 variant shows more frequent prolactinoma and less frequent gastrinoma than typical MEN1. Gonadotrophic tumors are occurring infrequently but may cause ovarian hyperstimulation.
Nonfunctioning tumors, GH, or adrenocorticotrophic hormone (ACTH) producing tumors and mixed tumors are seen less frequently.
Pituitary tumors in patients with MEN1 syndrome tend to be larger, invasive, and more symptomatic, and they tend to occur in younger patients when they are the initial presentation of MEN1.
Symptomatology
Elevated levels of prolactin may cause amenorrhoea, galactorrhoea, and lack of libido in females and hypogonadism in males.
Acromegaly, caused by a GH-producing tumor, is observed in 3–6% of MEN1 patients. Patients present with enlarged hands or feet, coarse facial features, or soft-tissue growth. Patients with acromegaly have an increased risk of developing cardiovascular disease, colon polyps, and cancer. Acromegaly may be associated with other tumors.
Nonfunctioning pituitary adenomas may grow large without symptoms. Due to compression of the surrounding tissues by the expanding tumor, complaints of visual field defects, headache, or an impairment of other pituitary functions may develop ( Fig. 24.1 c).
Diagnosis
The diagnosis is confirmed by determining plasma levels of prolactin (prolactinoma), one mg dexamethasone suppression test, and midnight salivary cortisol (Cushing’s disease), or insulin-like growth factor I (IGF-I) and by an oral glucose tolerance test to demonstrate absence of suppression of GH production (acromegaly). Pituitary adenomas can be detected visually by MRI with gadolinium contrast.
Adrenal Tumors
About 20% of MEN1 patients develop adrenal tumors. These tumors are often detected early by imaging of the upper abdomen every two years. Like sporadic incidentalomas of the adrenals, these tumors usually do not produce hormones and are mostly benign. Adrenal medulla tumors in MEN1 occur infrequently.
Neuroendocrine Tumors of Thymus, Lungs, and Stomach
In MEN1, NETs arise from cells that are derived from the embryonic foregut. NETs in MEN1 can develop in the thymus (mostly in males), in the lungs, and in the stomach, duodenum, or the pancreas (PETs).
They do not cause symptoms until at an advanced stage. As these tumors are capable of infiltrating surrounding tissues and metastasizing, and treatment is very difficult, early detection of these tumors is of vital interest.
NETs produce a vast spectrum of amines, peptides, and prostaglandins. NETs in MEN1 do not release serotonin (5HT), but do produce 5-hydroxytryptophan (5-HTP), the precursor of serotonin. The 5-HTP is partially converted to serotonin in the kidneys. Levels of platelet serotonin and chromogranin A are useful markers. The level of 5-hydroxyindoleacetic acid in the 24-h urine of MEN1 patients with NETs usually is not elevated.
Imaging
Tumors can be detected using MRI or CT scan, somatostatin receptor scintigraphy, or Ga-68-DOTATATE scintigraphy and endoscopy.
Thymus
Thymic NETs in MEN1 are associated with a very high lethality. Nearly all thymus carcinoid patients are male and smokers. Therefore, prophylactic thymectomy should be considered at neck surgery for primary hyperparathyroidism in male MEN1 patients, especially for smokers.
In 22 separate MEN1 families with thymic carcinoids, all but two (91%) have mutations coding for a truncated menin protein. There is clearly a high prevalence of truncating mutations in MEN1-related thymic carcinoids. Although when compared with the prevalence of truncating mutations among all reported MEN1 gene mutations, it is not significantly higher in MEN1 families with thymic NETs ( P = 0.39). In Japan, female patients are not rare exceptions.
Screening every patient affected with a neuroendocrine thymic neoplasm for MEN1 syndrome is recommended. In some families, an association with smoking occurs in approximately 25%. Khoo found a thymic NET in a father and son with MEN1.
Cushing’s syndrome due to ACTH-producing thymic carcinoid should also be considered as one phenotype of the MEN1 spectrum.
In a 761-patient MEN1 cohort from the Groupe des Tumeurs Endocrines registry, the probability of occurrence was 2.6% (range 1.3–5.5%) at age 40 years; seven patients (33%) belonged to clustered MEN1 families. Several aims, objectives were suggested for future guidelines: (1) earlier detection of Th-NET in MEN1 patients is required; (2) screening of both sexes is necessary; (3) a prospective study comparing MRI versus CT scan in yearly screening for Th-NET is needed; (4) a reinforced screening program must be established for patients who belong to clustered families; and (5) thymectomies must be performed in specialized centers.
Lung (NETs)
Bronchopulmonary NETs are relatively uncommon, occurring in approximately 3–8% of MEN1 patients. Multiplicity seems to be common in MEN-related lung NET. Very little is known of the natural course and prognosis of pulmonary carcinoids in MEN1. In one study, hypergastrinemia was significantly more common in patients with pulmonary nodules. No deaths or distant metastases occurred among these patients despite long-term follow-up. They did not appear to predict a poor prognosis in the majority of affected patients.
Gastric (NETs)
In MEN1 patients, gastric NETs have their origin in enterochromaffin-like (ECL) cells. Longstanding tumors may become symptomatic and demonstrate aggressive growth. Patients may have hypergastrinemia and ZES. With increased long-term medical treatment and life expectancy, these tumors will become an important determinant of survival. They require surgical treatment before they metastasize to the liver. Signet ring cell carcinomas may develop by gradual dedifferentiation from ECL cells (see Fig. 24.2 ).
Genetic pathophysiology of MEN1
Known Mutations and Specific Phenotypes
MEN1 is caused by inactivating germline mutations of the MEN1 gene, which is located on chromosome 11 (11q13). The MEN1 gene is a tumor suppressor gene. Biallelic inactivation of the MEN1 gene is required for the development of a tumor cell. Loss of the wild-type allele (loss of heterozygosity or LOH) is observed frequently in MEN1-associated tumors in MEN1 patients.
Since the discovery of the gene, more than 400 (459) different germline mutations have been identified in MEN1 families. These mutations are found scattered throughout the gene.
Also in sporadic MEN1-associated tumors, mutations of the MEN1 gene have been identified, which suggests that inactivation of the MEN1 gene contributes to the development of these tumors.
No clear genotype-phenotype correlation has been established. The expression of the disease is variable, even within families. However, some MEN1 gene mutations seem to be causing FIHP or a variant MEN1 that is characterized by the frequent occurrence of prolactinoma. Thus, additional genetic and/or epigenetic events may play a role in MEN1-associated tumorigenesis.
Pathophysiology of Mutations (How They Cause the Disease)
Normal Function of the Intact MEN1 Gene Product
The MEN1 gene encodes the menin protein. Menin is expressed ubiquitously, and performs its tasks predominantly in the nucleus (see Fig. 24.3 and Table 24.2 ).
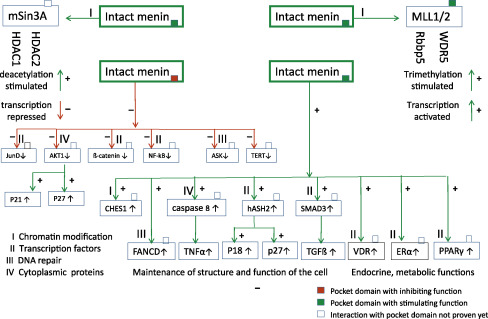
- •
corepressor on transcription of other target genes, for example, Jun D and hTERT
- •
coactivator on transcription of target genes, for example, P18, P27 and pS2
- •
corepressor function on a.o. JunD/c-Jun or hTERT is defective
- •
coactivator function on a.o. P18 and P27 is defective
| A. Chromatin modification | |
| Histone deacetylation | Histone protein preservation |
| Histone trimethylation | Histone protein destabilization |
| B. Transcription regulation | |
| Embryonic development | Homeobox genes |
| Inhibition of cell proliferation | p18, p27 |
| Apoptosis | Telomerase repression (hTERT) |
| Nuclear receptors for endocrine & | Steroid hormone receptors (ER, PR, GR, AR) |
| Metabolic functions | Vitamin D receptor (VDR) |
| Fatty acids (PPARγ) | |
| C. DNA-damage repair and stability | CHES1, FANCD2, RPA2, ASK |
| D. Cytoskeleton organization and cytoplasmatic processes | Vimentin Myosin |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


