Early diagnosis of multiple endocrine neoplasia (MEN) syndromes is critical for optimal clinical outcomes; before the MEN syndromes can be diagnosed, they must be suspected. Genetic testing for germline alterations in both the MEN type 1 (MEN1) gene and RET proto-oncogene is crucial to identifying those at risk in affected kindreds and directing timely surveillance and surgical therapy to those at greatest risk of potentially life-threatening neoplasia. Pancreatic, thymic, and bronchial neuroendocrine tumors are the leading cause of death in patients with MEN1 and should be aggressively considered by at least biannual computed tomography imaging.
Key points
- •
Early diagnosis of the multiple endocrine neoplasia (MEN) syndromes is critical for optimal clinical outcomes; before the MEN syndromes can be diagnosed, they must be suspected.
- •
Genetic testing for germline alterations in both the MEN type 1 (MEN1) gene and RET proto-oncogene is crucial to identifying those at risk in affected kindreds and directing timely surveillance and surgical therapy to those at greatest risk of potentially life-threatening neoplasia.
- •
Pancreatic, thymic, and bronchial neuroendocrine tumors are the leading cause of death in patients with MEN1 and should be aggressively considered by at least biannual computed tomography imaging.
- •
Patients with MEN-2a or 2b who are from a kindred and have a RET gene mutation identified should undergo total thyroidectomy after a pheochromocytoma is excluded.
Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 1 (MEN1) is inherited as an autosomal dominant disorder. It has a prevalence of 2 to 3 per 100,000 and is reported to be present in 0.22% to 0.25% of autopsies. The gene causing MEN1 is located on the long arm of chromosome 11 (11q13) and is composed of 10 exons (9 coding). The MEN1 gene is a tumor suppressor gene. It encodes a 610 amino acid nuclear protein called menin. Abnormalities of this gene result in mutations, deletions, and/or truncations of the menin protein. The exact mechanism by which alterations of menin result in endocrine tumors is still unclear. Menin interacts with several proteins, many of which have important roles in transcriptional regulation, genomic stability, cell division, and cell cycle control. The crystal structure of menin supports its role as a key scaffolding protein that regulates gene transcription.
Patients with MEN1 usually develop primary hyperparathyroidism (HPT) as the initial disorder of the syndrome (90%–100%), followed by pancreatic neuroendocrine tumors (pNETs) that can either be functional (20%–70%), of which gastrinoma is the most common, or nonfunctional (80%–100%) (pituitary adenomas [20%–65%], adrenal tumors [10%–73%], and thyroid adenomas [0%–10%]). Patients with MEN1 also have a high occurrence of other endocrine and nonendocrine tumors, including carcinoid tumors (thymic 0%–8%, gastric 7%–35%, bronchial 0%–8%, and rarely intestinal), skin and subcutaneous tumors (angiofibromas 88%, collagenomas 72%, lipomas 34%, and melanoma), central nervous system tumors (meningiomas, ependymomas, and schwannomas 0%–8%), and smooth muscle tumors (leiomyomas and leiomyosarcomas 1%–7%). In early studies, thyroid disease was also reported in patients with MEN1; but in a recent cross-sectional study of 95 patients with MEN1, the rate of co-occurrence of a thyroid incidentaloma was the same as matched non-MEN1 patients (45% vs 51%), respectively. In addition, other nonendocrine malignant tumors are also being reported to occur in MEN1. These tumors include lymphomas, renal cell cancer, melanoma, leiomyosarcoma, thrombotic thrombocytopenia purpura, myeloma, ovarian tumors, gastrointestinal stromal tumor, seminoma, chondrosarcoma, mesothelioma, and thymomas. However, whether the incidence of these nonendocrine tumors is truly increased or not is unclear.
Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 1 (MEN1) is inherited as an autosomal dominant disorder. It has a prevalence of 2 to 3 per 100,000 and is reported to be present in 0.22% to 0.25% of autopsies. The gene causing MEN1 is located on the long arm of chromosome 11 (11q13) and is composed of 10 exons (9 coding). The MEN1 gene is a tumor suppressor gene. It encodes a 610 amino acid nuclear protein called menin. Abnormalities of this gene result in mutations, deletions, and/or truncations of the menin protein. The exact mechanism by which alterations of menin result in endocrine tumors is still unclear. Menin interacts with several proteins, many of which have important roles in transcriptional regulation, genomic stability, cell division, and cell cycle control. The crystal structure of menin supports its role as a key scaffolding protein that regulates gene transcription.
Patients with MEN1 usually develop primary hyperparathyroidism (HPT) as the initial disorder of the syndrome (90%–100%), followed by pancreatic neuroendocrine tumors (pNETs) that can either be functional (20%–70%), of which gastrinoma is the most common, or nonfunctional (80%–100%) (pituitary adenomas [20%–65%], adrenal tumors [10%–73%], and thyroid adenomas [0%–10%]). Patients with MEN1 also have a high occurrence of other endocrine and nonendocrine tumors, including carcinoid tumors (thymic 0%–8%, gastric 7%–35%, bronchial 0%–8%, and rarely intestinal), skin and subcutaneous tumors (angiofibromas 88%, collagenomas 72%, lipomas 34%, and melanoma), central nervous system tumors (meningiomas, ependymomas, and schwannomas 0%–8%), and smooth muscle tumors (leiomyomas and leiomyosarcomas 1%–7%). In early studies, thyroid disease was also reported in patients with MEN1; but in a recent cross-sectional study of 95 patients with MEN1, the rate of co-occurrence of a thyroid incidentaloma was the same as matched non-MEN1 patients (45% vs 51%), respectively. In addition, other nonendocrine malignant tumors are also being reported to occur in MEN1. These tumors include lymphomas, renal cell cancer, melanoma, leiomyosarcoma, thrombotic thrombocytopenia purpura, myeloma, ovarian tumors, gastrointestinal stromal tumor, seminoma, chondrosarcoma, mesothelioma, and thymomas. However, whether the incidence of these nonendocrine tumors is truly increased or not is unclear.
Early diagnosis of multiple endocrine neoplasia type 1
Before MEN1 can be diagnosed, it must be suspected. Suspicion should be raised in any patient with a family history of endocrine tumors of the pancreas, family members with pituitary or parathyroid disease, or a family history of endocrinopathy; in patients with renal colic with NETs; in any patient with Zollinger-Ellison syndrome (ZES) (20%–25% have it as part of the MEN1 syndrome); with a young age onset of a functional pNET; with multiple pNETs; with HPT with multiple gland involvement or with hyperplasia or with a pNET associated with hypercalcemia or another endocrinopathy. In most patients (83%), MEN1 clinically presents after 21 years of age. In the 17% of patients with MEN1 presenting at less than 21 years of age, which should lead to suspicion of the diagnosis, the most frequent abnormalities were HPT (75%), pituitary adenoma (34%), insulinoma (12%), nonfunctional pNET (9%), and gastrinoma (2%). Genetic screening for MEN1 is recommended when an individual has 2 or more MEN1–related tumors, multiple abnormal parathyroid glands before 30 years of age, recurrent HPT at a young age, gastrinoma and HPT or multiple pNETs at any age, plus a family history of kidney stones or endocrine tumors that are part of the syndrome. Genetic testing includes sequencing of the entire coding region of the MEN1 gene (exons 2–10) and identifies mutations in about 80% of patients with familial MEN1 ( Table 1 ).
| Familial Endocrine Syndrome | Chromosome | Gene | Pattern of Inheritance | Glands Involved | Extraglandular Manifestations | Cause of Death |
|---|---|---|---|---|---|---|
| MEN1 | 11 | Menin | Autosomal dominant | Parathyroid, pancreas, pituitary, thyroid, adrenal | Bronchus, thymus, stomach, lipomas, skin tumors | pNET, bronchial or thymic NET |
| MEN-2a | 10 | RET | Autosomal dominant | Thyroid, adrenal, parathyroid | None | MTC |
| MEN-2b (MEN-3) | 10 | RET | Autosomal dominant | Thyroid, adrenal | Marfanoid habitus, mucosal neuromas, corneal nerve hypertrophy, intestinal ganglioneuromatosis | MTC |
| FMTC | 10 | RET | Autosomal dominant | Thyroid | None | Natural causes |
| MEN-4 | 12 | CDKN1B/p27Kip1 | Autosomal dominant | Parathyroid, GEP, pituitary | None | Unclear |
Parathyroid disease in multiple endocrine neoplasia type 1
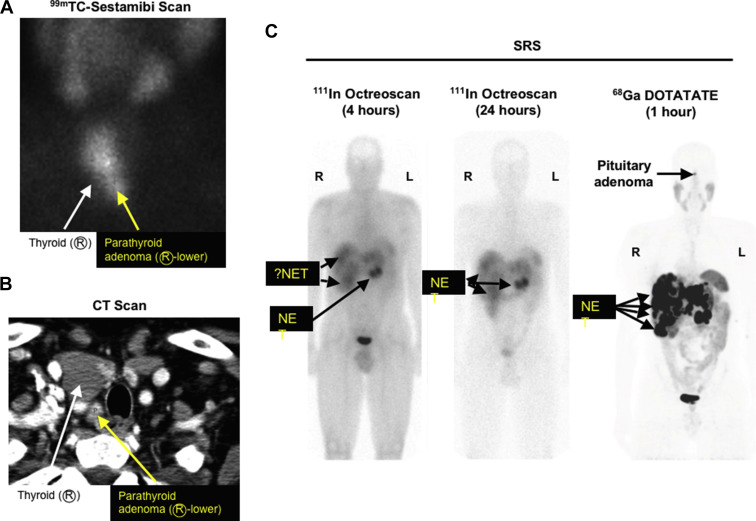
Primary HPT is the most common endocrine abnormality in MEN1. It reaches nearly 100% penetrance by 50 years of age. HPT is usually the first manifestation of MEN1 with a typical age of onset of 20 to 25 years. Decreased bone density and kidney stones are common. HPT often occurs at the same time as ZES and surgery to correct the HPT greatly ameliorates the clinical findings of ZES. As in sporadic cases, biochemical testing for HPT is critical to the diagnosis. Total or ionized serum level of calcium and intact serum parathyroid hormone levels are measured and both should be elevated. 24 hour urinary calcium should also be measured and will be elevated. Opinions differ as to the timing of parathyroid surgery in MEN1 as well as to the type of operation that should be performed. Early surgery may delay the lifetime exposure to the biochemical manifestations of HPT, whereas waiting until the parathyroid glands enlarge may make the operation easier. Patients with MEN1 and HPT typically have multiple abnormal glands. The tumors are asymmetric in size and should be considered as independent clonal adenomas. Imaging studies are not useful for initial operations because all 4 parathyroid glands must be identified but are necessary and very useful in reoperations ( Fig. 1 ). Some have recommended in patients with MEN1 a minimally invasive parathyroidectomy should be considered with selective removal of only the enlarged glands, whereas others recommend subtotal parathyroidectomy (3.5 glands removed) or a total parathyroidectomy with inplant. The current operation that is recommended by most experts is a subtotal parathyroidectomy (3.5 gland resection) with removal of the cervical thymus. Intraoperative parathyroid hormone level monitoring is recommended to be certain that sufficient abnormal parathyroid tissue has been removed. A viable 50-mg amount of normal parathyroid tissue should be left in the neck and marked with a hemoclip. Total parathyroid resection and transplantation of parathyroid tissue to the nondominant forearm used to be recommended, but it has been shown to have too high an incidence of hypoparathyroidism caused by graft failure. Because of the multiple abnormal parathyroid gland nature of this disease, there is a high probability of recurrent HPT years after surgery if less than 3.5 parathyroid gland resections are performed. Patients should be followed for this possibility. Calcium-sensing receptor agonists (calcimimetics) are a new class of drugs that can act directly on the parathyroid gland, decrease parathyroid hormone release, and may even decrease parathyroid tissue growth. A small number of patients with MEN1 and HPT have been treated with the calcium-sensing receptor agonist cinacalcet, and it has been effective in the control of the HPT. These agents may play an important role in the management of these patients in the future. Parathyroid carcinoma has been reported in patients with MEN1 ; however, it is uncommon, occurring in only 0.28% of all patients with MEN1. The clinical presentation is similar to that seen in patients without MEN1 with parathyroid carcinoma. Patients with MEN1 commonly develop bone disease with osteoporosis thought secondary to the HPT. However, recent studies show that menin has important effects in bone particularly in regulating osteoblast activity, which plays a key role in bone development, remodeling, and maintenance. Some patients with MEN1 require multiple reoperations to control the recurrent or persistent HPT, and reoperation can be difficult and associated with increased morbidity. Recently ethanol ablation of abnormal parathyroids has been described to be able to successfully control the HPT in such patients with MEN1 with a low rate of hypocalcemia and no permanent complications.
Pituitary tumors in multiple endocrine neoplasia type 1
Anterior pituitary adenomas are the initial clinical manifestation of MEN1 in 25% of cases. Its prevalence in MEN1 is between 20% and 60%; in a recent study of all patients (N = 144) with pituitary adenomas seen in one institution over a 6-year period, 7.7% had MEN1. Most of these anterior pituitary tumors are microadenomas (<1 cm in diameter). Every type of anterior pituitary tumor has been reported to occur in MEN1, with the most common being a prolactinoma. Screening for anterior pituitary tumors requires measuring serum levels of prolactin and insulinlike growth factor 1 and MRI of the pituitary. Patients should be questioned for loss of peripheral vision. Visual fields are assessed formally if any suspicion of change or there is evidence of a pituitary tumor. Because these tumors occur in patients of childbearing age, undiagnosed pregnancy may cause a confusingly elevated prolactin level. Treatment of pituitary tumors in MEN1 is the same as sporadic pituitary tumors. Bromocriptine and cabergoline are used to treat prolactinomas. Octreotide and lanreotide are used to treat growth hormone–secreting tumors. Transsphenoidal surgery is the treatment of choice for discrete pituitary microadenomas or macroadenomas and may be curative. The major surgical morbidity of transsphenoidal hypophysectomy is permanent diabetes insipidus. Even in patients who are successfully treated, long-term follow-up is indicated because tumors may recur.
Pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1
The prevalence of pNETs in MEN1 is between 30% and 75% clinically and between 80% and 100% in postmortem studies. The pathologic findings of pNETs in MEN1 is typically multicentric and multifocal with multiple endocrine tumors throughout the pancreas and the duodenum in patients with MEN1/ZES ( Fig. 2 ). Unfortunately the only reliable method of establishing the presence of pNETs in patients with MEN1 not associated with a clinical syndrome (nonfunctional pNETS [NF-pNETs]) is to perform detailed imaging studies. Although NF-pNETs can secrete several peptides, including chromogranin A, neuron-specific enolase, pancreatic polypeptide, neurotensin, or ghrelin, these do not result in a distinct clinical syndrome; a recent study demonstrates their assessment in plasma in patients with MEN1 have low diagnostic accuracy for NF-pNETs. The frequency with which pNETs are detected by imaging depends to a large extent on the imaging modality. Cross-sectional imaging studies (computed tomography [CT], MRI, transabdominal ultrasound) frequently (35%) miss tumors less than 2 cm; indium In 111 pentetreotide (Octreoscan) misses 25% to 30% less than 1 cm; gallium-68 ( 68 Ga)–DOTATOC/PET misses 20% to 30% less than 0.5 cm; and endoscopic ultrasound (EUS) can detect pNETs down to 0.1 to 0.2 cm. 68 Ga-DOTATE is less available than Octreoscan but is more sensitive and frequently images more tumors than Octreoscan (see Fig. 1 ). Studies in patients with MEN1 show that EUS is the most sensitive single modality to detect pNETs. In one recent comparative study, EUS was markedly superior to Carbon 11 5-Hydroxytryptophan scanning, somatostatin receptor scintigraphy (SRS), and CT/MRI for the detection of pNET. However, in another recent prospective study, the findings of MRI and EUS were complementary in patients with MEN1 for the detection of pNETs. Tumors vary from microadenomas to adenomas to carcinomas (see Fig. 2 ) with lymph node and liver metastases. The most characteristic MEN1 pancreatic lesion on pathology studies is the presence of diffuse microadenomatosis (<0.5 mm). Molecular studies in multiple pNETs or gastrinomas from patients with MEN1 assessing loss of heterozygosity at various loci and X-chromosome inactivation studies demonstrate that the multiple pNETs arise as independent events. Although microadenomatosis of the pancreas is almost invariably seen, in various studies, only 0% to 13% of patients with MEN1 are reported to develop large (>2 cm) and/or symptomatic NF-pNETs, with many patients not developing larger pNETs even over long periods of time. At present the molecular events that lead to this variability in growth of pancreatic microadenomas in different patients with MEN1 are unknown, and there are well-established predictive factors requiring sequential imaging as the only means to evaluate the growth over time. Most studies in patients with MEN1 show no genotype-phenotype correlations ; however, a few recent studies have reported various MEN1 gene mutations more frequently in patients with aggressive tumors or with decreased survival; but they are not widely used or generally established for clinical use. These mutations include inactivating mutations of the MEN1 gene; the presence of exon 2 or nonsense/frameshift mutation in exon 2, 9, and 10; the presence of mutations affecting the June D interacting domain; and the presence of mutations affecting the checkpoint kinase 1 interacting domain. Islet cell hyperplasia is rare. Duodenal gastrinomas in MEN1 are usually small (<1 cm), submucosal, multifocal, and, as seen in sporadic ZES, occur primarily in the proximal duodenum. Lymph node metastases occur in 45% to 95% of both duodenal and pancreatic gastrinomas, demonstrating that they are equally malignant; however, liver metastases occur less frequently in patients with duodenal NETs than pNETs, demonstrating that they are not equally aggressive. pNETs contain, in decreasing frequency, chromogranin A, pancreatic polypeptide, glucagon, insulin, proinsulin, somatostatin, gastrin, vasoactive intestinal polypeptide, serotonin, calcitonin, growth-hormone releasing factor, and neurotensin. Just because tumors may stain positive for these hormones by immunohistochemistry does not mean that they are associated with a clinical syndrome. A functional pNET, by definition, demonstrates excessive secretion of hormones associated with symptoms of hormonal excess. Malignant pNETs are rare before 30 years of age; however, 50% of middle-aged patients with MEN1 have evidence for a malignant pNET. A prospective study in patients with MEN1/ZES demonstrated that 14% of all patients had pNETs/gastrinomas demonstrating aggressive growth that was associated with decreased survival, which is a lower rate than seen in patients with sporadic ZES, wherein 25% are reported to show an aggressive growth pattern.
One of the most significant controversies concerning patients with MEN1 is the role of surgery in the management of various aspects of pNETs. Almost all agree that surgical resection should be undertaken for MEN1 patients who develop insulinomas (18%), or other rare functional pNETs, such as VIPomas, glucagonomas, Growth Hormone Releasing Factor (<5%), or very rarely functional somatostatinomas (<1%). However, there is no agreement on surgery’s role in patients with the most frequent functional pNET, gastrinomas causing the Zollinger-Ellison syndrome, that occur in 54% of patients or NF-pNETs that develop in 80% to 100% of patients. There are several reasons for this controversy. In contrast to patients with insulinoma or the other rarer functional pNETs, in which surgery is frequently curative, surgery for patients with MEN1 with ZES or with NF-pNETs is seldom curative. Therefore, surgical interventions must be carefully considered in a true risk-benefit analysis. This risk-benefit ratio is difficult to calculate for several reasons. A primary factor is that the natural history of patients with ZES, pNET, and MEN1 is changing and largely unknown ( Fig. 3 ). In early series, before effective medical control of gastric acid hypersecretion caused by ZES existed, most patients with MEN1 died of the complications of the uncontrolled acid hypersecretion ( Fig. 4 ). With the development of effective medical therapies (histamine H2-receptor antagonists [1970–80s] and proton pump inhibitors [PPIs] [1985-present]), the gastric acid secretion became an uncommon cause of death (<20%) (see Fig. 4 ). In contrast, death from the malignant behavior of an NET has increased in frequency to greater than 60% in recent series (see Fig. 4 ). Patients with MEN1 are now living longer (mean age at death: 55–60 years) (see Fig. 3 ), and the death rate from other malignant tumors is increasing. This finding is particularly true of thymic NETs (carcinoids). Currently, thymic carcinoids are the cause of death in approximately 10% to 25% of patients with MEN1. These carcinoids are the most lethal NETs in MEN1. Further, a wide range of other tumors are increasingly reported in patients with MEN1. Even though malignant NETs are reported as a cause of death of patients with MEN1 in various series, the exact source of the metastatic NET is usually not clearly established. This point is illustrated by the fact that a given patient may have a pNET and a carcinoid tumor (gastric, thymic, or pulmonary), so the source of the metastatic disease is unclear. This consideration is further complicated by the fact that small NF-pNETs have an excellent long-term survival ; in fact, their survival is not different from patients without pNETs. An additional factor affecting the role of surgery is the fact that patients with MEN1 and pNETs may have life-threatening symptoms secondary to unregulated hormonal overproduction of the tumor. Examples include peptic ulcer disease and diarrhea secondary to gastrinoma, hypercortisolism secondary to ectopic ACTH secretion, and hypoglycemia secondary to an insulinoma. In the past, surgery was frequently the only hope to ameliorate and control these symptoms. However, effective drugs can control these symptoms, making surgery less necessary. Specifically, PPIs can control acid hypersecretion in all patients with MEN1/ZES ; long-acting somatostatin analogues can control severe diarrhea in VIPoma and necrolytic migratory pruritic rash in glucagonoma ; diazoxide can control hypoglycemia in insulinoma. Lastly, surgery may be associated with both short-term and long-term complications that may result in morbidity and mortality. For example, some studies suggest that patients with MEN1 have an increased incidence of diabetes mellitus and that pancreatic resections exacerbate this condition. Furthermore, because microscopic pNETs are almost invariably left in the pancreas, curative surgery may require a total pancreatectomy, which can result in a greatly impaired quality of life and a difficult management course. Each of these factors (unknown natural history, low cure rate without aggressive surgery, excellent prognosis for small pNETs in MEN1/ZES-NF-pNETs, good medical control of functional pNETs, short-term/long-term surgical side-effects) has made it difficult to calculate the true risk/benefit ratio of surgery for pNETs in patients with MEN1. There are currently no clear markers that identify the cases that are at greatest risk for progression and death caused by a malignant pNET. In recent studies, several clinical/laboratory/tumoral features are reported to have prognostic value in patients with MEN1. These features include the presence of thymic carcinoids, the presence of more than one functional hormonal pNET syndrome, the need for greater than 3 parathyroid surgical procedures, the presence of either liver metastases or distant metastases from the pNET, aggressive primary tumor growth (invasion into superior mesenteric vein, bile duct obstruction), large pNETs (>4 cm), pNETs with areas of poor vascular enhancement on CT, calcifications on CT, and serial imaging with evidence for progression by increasing tumor size or new lesions.
Gastrinomas (ZES) are the most common functional pNET in MEN1. Recent studies show the duodenal but not pancreatic gastrinomas develop by hyperplasia of duodenal G cells. With increasing hyperplasia and subsequent loss of heterozygosity (LOH) at the 11q13 MEN1 locus, duodenal microgastrinomas arise. ZES is diagnosed by demonstrating an elevated fasting serum level of gastrin (off PPIs) with a concomitant increased gastric acid output (>10 mEq/h). Correlates for a poor prognosis with MEN1/ZES are pancreatic tail primary tumors, hepatic metastases, tumors that make both gastrin and adrenocorticotropic hormone (ACTH), distant metastases, and severe hypergastrinemia (defined as a level >3000). Various surgical approaches have been performed in an attempt to cure patients with MEN1/ZES. However, patients with MEN1 typically have multiple, small duodenal gastrinomas associated with lymph node metastases in greater than 50% of cases, in addition to multiple pNETs, which are uncommonly the cause of the ZES (<15%) (see Fig. 2 ). Almost all studies show that local removal of duodenal gastrinomas is associated with persistent ZES. However, Whipple pancreaticoduodenectomy has been reportedly associated with biochemical cure. This latter approach is controversial because the symptoms of ZES are well controlled with PPIs; studies show patients with MEN1/ZES have an outstanding long-term survival on PPIs (up to 100% at 15 years); and the immediate and long-term morbidity/mortality of the Whipple procedure may be too great. An argument for the more aggressive surgical procedure is that with even longer follow-up a higher percentage of these patients with MEN1/ZES develop malignant gastric carcinoid tumors that may also affect survival ( Fig. 5 B). Therefore, the surgical approach to these patients will likely remain controversial until more data on the natural history becomes available. The authors’ approach, because of its excellent quality of life and outlook, is to currently recommend only local resection (excision of duodenal tumors and/or enucleation of pancreatic head tumors with lymph node sampling and distal pancreatectomy for body/tail tumors that are >2 cm). Lymph node sampling is recommended because it has prognostic value and perhaps can increase the cure rate. Although uncommonly curative, it is associated with acceptable morbidity and mortality and long-term tumor control and survival with a high quality of life. If patients have larger (>2.0–2.5 cm) pancreatic head tumors with positive lymph nodes, then the authors agree that more radical resection (proximal pancreaticoduodenectomy) may be considered in selected cases, although the authors’ usual operation is to locally resect these tumors and not to perform a pancreaticoduodenectomy. The authors’ recommendations are consistent with those of both large neuroendocrine tumor societies’ guidelines recently published (ie, the North American Neuroendocrine Tumor Society [NANET] and the European Neuroendocrine Tumor Society [ENET] ) as well as the clinical practice guidelines for MEN1 published by the Endocrine Society.
Insulinoma is the second most common functional pNET in MEN1. Approximately 10% to 18% of patients with MEN1 will develop insulinoma, and 5% to 10% of insulinomas occur in the setting of MEN1. Insulinoma is the first manifestation of MEN1 in up to 10% of patients. Patients have hypoglycemia and neuroglycopenic symptoms (altered mental status and seizures). This manifestation characteristically occurs at a young age (<35 years). Fasting hypoglycemia (glucose <45 mg/dL) and concomitant hyperinsulinemia (levels >5 uU/mL) are diagnostic. Insulinomas are generally small (<2 cm) and distributed uniformly throughout the pancreas. Because patients with MEN1 have multiple pNETS, it may not be clear which tumor is secreting the excessive insulin. However, studies have reported that the insulinoma in MEN1 is most commonly a dominant (>2 cm) pNET that is frequently identified by conventional imaging studies like CT or MRI, usually in the body and tail of the pancreas. Medical control of the hypoglycemia is not as effective as that with other functional tumors making surgery more important for symptom relief. Insulinomas in the pancreatic body and tail are removed by a distal pancreatectomy. Tumors in the head are enucleated. Preoperative endoscopic and intraoperative ultrasound can provide precise localization of the tumor and may facilitate excision by imaging the relationship to the pancreatic duct. Patients with MEN1 with an insulinoma and no imaging evidence of a dominant tumor should undergo calcium angiogram. This angiogram will localize the section of the pancreas containing the insulinoma. Surgical resection can usually be done laparoscopically if the lesion is well localized preoperatively. Patients need to be followed for subsequent development of recurrent tumor and hypoglycemia. In a recent study of 73 patients with MEN1 with insulinoma after distal pancreatectomy (63%), total/cephalic pancreatectomy (12%), or enucleations (25%), at a median follow-up of 9 years, 82% remained hypoglycemia free. The recurrence rate was higher with enucleation, but the long-term complication rate was much greater (43%–55%) with resections than with enucleation (0%) ( P = .002). This finding led to the conclusion that, although resection is associated with a lower recurrence rate, enucleation alone may be an alternative because of its lower long-term morbidity.
NF-pNETs are a frequent problem in patients with MEN1, occurring as microadenomas in 80% to 100% on pathology examination (see Fig. 2 ), in 25% to 60% by imaging studies, and causing symptoms in up to 12% in different studies. For NF-pNETs in asymptomatic patients with MEN1, there is controversy over the role of surgery, the timing of surgery, and the type of surgery performed. Studies have demonstrated that patients with small NF-pNETs (<2 cm) have an excellent long-term survival; in fact, it is the same as patients with MEN1 without any pNET. Several groups recommend avoiding surgery if the NF-pNET is less than 2 cm or slowly growing. Others recommend surgery for tumors that are 1 cm. Still others recommend routine surgery for all patients in whom a pNET is identified by any method, even if it is identified biochemically and not imaged. Another approach is to perform serial EUS on patients with small pNETs (<1–2 cm) and operate if growth occurs. At present there is no consensus as to what size or change in size should be used for intervention. This lack of consensus is caused in large part by a lack of information on the natural history of NF-pNETs, especially those less than 1.5 to 2.0 cm in size that are asymptomatic. Recently several studies have attempted to address this question by following patients with MEN1 with serial EUSs that have been shown to be reliable for assessing changes in pNET size and to be able to detect additional small (<0.5 cm), new NF-pNETs as well as changes in existing NF-pNETs. These studies show that most NF-pNETs less than 1.5 to 2.0 cm remain stable or even decrease in size, with most of the rest only increasing in size slowly. At present too few patients have been studied in this manner to allow specific criteria about what rate of change or other alteration in the NF-pNET should lead to surgical intervention. It has been suggested that if change occurs, biopsy may help identify which patients should undergo surgery. The goal of surgery in MEN1 with NF-pNETs is to control tumor growth and prevent progression. Recent studies identify the following factors as suggestive of poor prognosis: higher fasting serum levels of gastrin, presence of more than one functional hormonal syndrome, need for greater than 3 parathyroid surgical procedures, presence of distant metastases from pNET, aggressive primary tumor growth (invasion into the SMV, bile duct obstruction), large pNETs (>4 cm), pNETs with areas of poor vascular enhancement on CT, calcifications, and imaging evidence of progression. EUS and fine-needle or core-needle biopsy can be especially useful for determining the malignant potential by measuring the Ki67 rate (see Fig. 2 ). Further, 68 Ga-DOTATOC PET/CT can be used to better stage the true extent of tumor in patients with MEN1. It is very useful because it is a sensitive whole-body study; it may identify other primary NETs like pituitary, bronchus, thymus, stomach, and ileum. It is more sensitive than Octreoscan, which uses 111 In-labeled pentetreotide with single-photon emission computed tomography imaging (see Fig. 1 ). 68 Ga-DOTATOC PET/CT was originally available only in Europe, and now it is becoming available in the United States. The standard operation for NF-pNETs is distal or subtotal pancreatic resection with intraoperative ultrasound and enucleation of tumors from the pancreatic head and duodenum. For bulky (>2.0–2.5 cm) tumors in the head of the pancreas, proximal pancreaticoduodenectomy or the Whipple procedure may be necessary. Dissection of lymph nodes along the celiac axis and hepatoduodenal ligament is also indicated. Extensive pancreaticoduodenal resection is associated with an increased risk and is primarily indicated for clearly malignant tumors, as these tumors must be controlled. At present, because these patients have excellent long-term survival (see Fig. 3 ), the authors recommend surgical exploration only in patients with MEN1 with NF-pNETs greater than 2 cm or symptomatic. This recommendation is consistent with the guidelines by both large neuroendocrine tumor societies (ie, NANETs and ENETs ) as well as the clinical practice guidelines for MEN1 published by the Endocrine Society.
Management of rare functional pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1
Glucagonomas occur in 3% (range 1%–6%) of patients with MEN1, VIPomas in 3% (range 1%–12%), and GRFomas and somatostatinomas in less than 1%. All guidelines agree that if unresectable disease is not present and patients have no medical contraindication to surgery, these patients should undergo surgical exploration for potential cure and control of the malignant nature of the pNET.
Stomach, thymic, and bronchial neuroendocrine tumors and adrenal cortical tumors
Patients with MEN1 develop type II gastric carcinoid tumors (7%–35%) (see Fig. 5 ), bronchial carcinoid tumors (0%–8%) ( Fig. 6 ), thymic carcinoid tumors (0%–8%), and adrenal cortical tumors (27%–36%, <2% symptomatic). In a recent study of patients with MEN1 who died, 45% had adrenal cortical tumors, 19% gastric carcinoid tumors, 10% lung carcinoid tumor, and 6% thymic carcinoid tumor. Of these less common tumors listed, the tumor that seems to cause death most frequently was the thymic carcinoid tumor (see Fig. 5 ). It was second only to pNETs as the cause of death in patients with MEN1. Unfortunately, thymic NETs in patients with MEN1 are rarely discovered when completely resectable; most present with advanced disease encasing great vessels, invading surrounding tissues, and frequently with bone and/or liver metastases. In contrast to thymic carcinoids occurring in a non-MEN1 setting, thymic carcinoids occurring in patients with MEN1 are rarely associated with ectopic hormone production, such as Cushing syndrome. Early diagnosis by awareness and imaging with 68 Ga-DOTATE, CT, or MRI followed by complete surgical resection are the mainstays of treatment.
Type 2 gastric carcinoid tumors occur almost entirely in patients with MEN1 with ZES and are thought to be secondary to the trophic action of chronic hypergastrinemia on the gastric enterochromaffin-like (ECL) cells combined with the presence of LOH at the MEN1 locus on 11q13 (see Fig. 5 ). Studies show that almost all patients with MEN1/ZES have gastric ECL cell proliferative changes, which are currently thought to be precursor changes leading to the development of gastric carcinoids in these patients. A large prospective study found gastric ECL proliferative changes were present in all patients with MEN1/ZES studied; the changes were advanced in greater than 50% of the patients and more severe than that seen in patients with sporadic ZES without MEN1. The natural history is unclear because before the mid 1980s most patients with ZES underwent a total gastrectomy and, thus, did not develop these tumors. More recent studies show these type 2 gastric carcinoids (5%–6% all gastric carcinoids) are more aggressive than the more common type 1 found in patients without MEN1 with atrophic gastritis/pernicious anemia (70%–80% all gastric carcinoids). Type 2 metastasizes to the liver at a higher rate (10%–30%). Those with localized disease (>70%–90%) are usually excised endoscopically after assessing the extent of invasion by EUS; but in some patients, the tumors are present in excessive numbers and a larger size. In these latter patients, the gastric NETs can be invasive so that additional treatments may be needed. In some cases, aggressive surgical resection (subtotal or total gastrectomy and D-2 lymph node dissection) is recommended and additional treatment with long-acting somatostatin analogues or cholecystokinin B receptor antagonists is administered. Because most gastric carcinoids develop asymptomatically and because they can be more aggressive than type 1 gastric carcinoids that occur in atrophic gastritis, it is recommended that all patients with ZES/MEN1 undergo regular endoscopic evaluation (annually).
Curative resection (lobectomy with lymph node dissection) is recommended for bronchopulmonary NETs (see Fig. 6 ) and radical median sternotomy with thymectomy for thymic carcinoid tumors (see Fig. 5 ). Although most bronchopulmonary NETs can be completely resected, unfortunately this is not the case for most thymic carcinoids, which are very aggressive tumors and metastasize early, especially to bone. Thymic carcinoids were not recognized as part of the MEN1 syndrome until the 1980s, so there is only limited information on their natural history. Controversy exists as to the best method for their early detection, whether surgical resection should be performed even if distant metastatic disease is present, whether the recommendation of routine thymic resection during parathyroid surgery for HPT reduces the risk of the subsequent development of thymic carcinoids, and whether radiation or some other ant-tumor treatment should be given after surgical resection. Current guidelines recommend screening CT or MRI screening of the chest every 1 to 2 years and continued cervical thymectomy when parathyroid glands are removed; many recommend aggressive resection even with metastatic disease to prevent local complications.
Adrenal tumors in patients with MEN1 are frequent (27%–36%); however, they are usually small (<3–4 cm) (>80%), nonfunctional (85%), benign (>86%), and asymptomatic (>98%). These tumors may cause primary hyperaldosteronism and primary hypercortisolism and may become malignant. At present adrenal tumors in patients with MEN1 are generally treated as that for non-MEN1 adrenal tumors. Some studies suggest important differences in adrenal tumors in patients with MEN1 and non-MEN1 patients. At present the exact screening time, tumor size that should be appropriated for surgical intervention, and frequency of assessment of functionality have not been systematically studied and defined to detect early malignant change or functionality in patients with MEN1.
Management of metastatic disease in multiple endocrine neoplasia type 1
Of the MEN1–associated tumors, metastatic disease most commonly occurs with pancreatic, thymic, and bronchial NETs and occasionally in gastric carcinoid tumors. For example, approximately 60% of patients with duodenal gastrinomas or pNETs have metastatic disease at the time of diagnosis. The presence of liver metastases initially or their development subsequently is associated with a poor prognosis and decreasing survival. One of the unique problems confounding the treatment of patients with MEN1 is often the fact that the exact primary source of the liver metastases is unclear. It may be a pNET, a duodenal gastrinoma, thymic carcinoid, or another carcinoid like the stomach or bronchial. For example, there are several reports of patients with MEN1/ZES with liver metastases from a NET that is not the gastrinoma. Liver metastases in patients with MEN1 and a pNET and without apparent carcinoid tumors like thymic, pulmonary, or stomach are almost always attributed to the pNET. This ambiguity may be resolved because recently pathologists are using different immunohistochemical studies to try to determine the true primary source of the metastasis (ie, PAX8, TTF-1, CDX-2, and so forth). This distinction of the true source of the metastases is becoming more important because recent studies with several therapies used in patients with advanced metastatic NET disease (somatostatin analogues, everolimus, sunitinib, chemotherapeutic agents) demonstrate that pNETs and gastrointestinal (GI)-NETs (carcinoids) respond differently. Management of metastatic disease in patients with MEN1 is generally comparable with the management of patients without MEN1. However, for patients with multiple primary endocrine tumors, vigilance must be taken when evaluating new findings on cross-sectional imaging to be certain which tumor is metastatic and/or progressing. The oncologic management of metastatic NETs has improved with new drugs showing response rates primarily in pNETs. The Food and Drug Administration (FDA) has recently approved 2 agents for the treatment of metastatic pNETs, the mammalian target of rapamycin inhibitor everolimus and the tyrosine kinase inhibitor sunitinib. Both have shown promising antitumor effects in recent studies. For advanced GI-NETs, 2 double-blind, randomized, placebo-controlled somatostatin analogue studies (PROMID, CLARINET ) as well as other studies show both the somatostatin–long-acting analogue octreotide-LAR and lanreotide autogel have antiproliferative effects, respectively. In addition, other novel therapies are being evaluated for the treatment of advanced pNETs and GI-carcinoids, including the use of radiolabeled somatostatin analogues ( 90 yttrium, 177 lutetium-labeled analogues) using the finding that almost all well-differentiated GI-NETs overexpress at least one subtype of somatostatin receptor, which can be used to image as well as target these cytotoxic agents; this approach is currently undergoing phase III trials.
In summary, the importance of the authors’ initial comments regarding the timely diagnosis of MEN1 cannot be overstated. Again, before MEN1 can be diagnosed it must be suspected . Suspicion should be raised in any patient with a family history of endocrine tumors of the pancreas, family members with pituitary or parathyroid disease, or a family history of endocrinopathy; in patients with renal colic with NETs; in any patient with ZES (20%–25% have it as part of the MEN1 syndrome); with a young age onset of a functional pNET; with multiple pNETs; with HPT with multiple gland involvement or with hyperplasia or with a pNET associated with hypercalcemia or another endocrinopathy. Genetic screening for MEN1 is recommended when an individual has 2 or more MEN1–related tumors, multiple abnormal parathyroid glands before 30 years of age, recurrent HPT at a young age, gastrinoma and HPT or multiple pNETs at any age, plus a family history of kidney stones or endocrine tumors that are part of the syndrome.
Patients with MEN1 are living longer and less frequently dying from the hormonal effects of tumors. However, the potentially malignant nature of some tumors, like pNETs, bronchial NETs, and thymic NETs, are accounting for an increasingly high proportion of deaths. Additionally, there is a possibility that these patients may have a higher incidence of other malignant nonendocrine tumors. Awareness of these possibilities and the use of improved imaging modalities, like CT, MRI, EUS, SRS, and 68 Ga-DOTATOC PET/CT, should diagnose these tumors earlier and in a more treatable state.
Multiple Endocrine Neoplasia Type 4
Recently a new MEN1–related syndrome has been defined and recognized entitled MEN-4. It has long been known that in patients with the MEN1 syndrome clinically 70% to 85% were found to have mutations in the MEN1 gene with familial disease and 30% of patients with sporadic MEN1. In studies that also assess large deletions, the percentage with MEN1 mutations in patients with familial MEN1 can increase to 90%. Thus, a percentage of patients with MEN1 features seem to have the disease based on some other genetic alteration. Recently it has been established that germline mutations in the cyclin-dependent kinase (CDK) inhibitor gene CDKN1B is responsible for causing MEN-X, a syndrome in rats with features of both the human MEN1 and MEN-2 syndromes. Subsequently, several patients with MEN1 features without a MEN1 gene mutation have been described who have mutations in the CDKN1B gene. The CCDKN1B gene encodes for a member of the CDK inhibitor family, p27 (also called KIP1), a nuclear protein that is important in regulating the cell cycle, particularly the transition from the G1 to S phase. The CDKN1B gene, similar to the MEN1 gene, functions as a tumor suppressor gene. Alterations in other cyclin-dependent kinase genes have also been reported in patients with clinical MEN1 features but no MEN1 gene mutations. In a study of 196 patients with clear MEN1 or suspected MEN1 but no MEN1 gene mutation, the relative frequency of the various CDK mutations were p15 (1.0%), p18 (0.5%), p21 (0.5%), and p27 (1.5%). In other studies, it is estimated that 3% of patients with MEN1 features have CDK1B mutations.
Multiple Endocrine Neoplasia Type 2
Multiple endocrine neoplasia type 2 is composed of 3 distinct clinical subtypes: MEN-2a, MEN-2b, and familial medullary thyroid carcinoma (FMTC). MEN-2 is a rare syndrome with an incidence of 1 in 200,000 live births. Each subtype is an autosomal dominant familial cancer syndrome associated with a germline mutation of variable penetrance in the RET proto-oncogene. Because 50% of children of an affected parent will manifest MEN-2, the syndrome occurs in every generation of a family. The principle feature of all MEN-2 subtypes is medullary thyroid carcinoma (MTC), a cancer of the parafollicular calcitonin secreting C cells. Calcitonin and carcinoembryonic antigen (CEA) are sensitive and, in the case of calcitonin, specific blood markers for MTC. Patients with MEN-2 have a 100% risk of developing MTC by 70 years of age, and MTC is the most important determinant of mortality in these patients. If MTC has spread beyond the thyroid, the prognosis is poor. Further, patient survival is greatly improved when MTC is treated surgically early in the course of the disease. Therefore, the emphasis on treating patients with MEN-2 is screening and early detection of MTC. MEN-2 is subgrouped into 2 variants called MEN-2a and MEN-2b (sometimes called MEN-3). Common features of the 2 groups are multicentric, bilateral MTC, occurring in all patients and bilateral pheochromocytomas occurring in 50% of patients. MEN-2a is the most common manifestation of MEN-2, accounting for 55% of cases. MTC is often the first manifestation of MEN-2a, usually occurring between 20 and 30 years of age. MEN-2a is characterized by MTC and pheochromocytomas plus primary HPT apparent in 20% to 30% of patients and a normal physical appearance and body habitus.
MEN-2b is a more rare form, accounting for approximately 5% to 10% of all cases. MEN-2b is marked by an early onset of a more aggressive form of MTC. MTC develops within the first year of life, and patients die before the 30 years of age. Patients with MEN-2b do not have parathyroid disease but do have a characteristic appearance, including Marfanoid habitus, pectus abnormalities, mesodermal abnormalities, corneal nerve hypertrophy, labial and mucosal neuromas, and intestinal ganglioneuromatosis. FMTC is another hereditary endocrine syndrome. It is the second most common variant. It accounts for 35% of cases. FMTC is the mildest subtype of MTC, and patients usually do not die of MTC. Patients only have MTC without any of the other features seen in MEN-2a or 2b. The diagnosis of FMTC should be considered when at least 4 family members develop MTC without other endocrine findings.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


