Classic examples of chromosomal-mediated antibiotic resistance include genetic mutations of antimicrobial targets leading to resistance for rifamycins (via mutations of the
rpoB gene) (
29) and for fluoroquinolones (via mutations in DNA topoisomerase) (
30). Alteration of promoters of gene expression via chromosomal mutation, thereby changing the production level of antibiotic-inactivating enzymes, antimicrobial targets, or membrane influx or efflux systems, also can lead to resistance. For example, mutations in the promoter controlling the expression of the
OprD porin in
P. aeruginosa can block the entry of carbapenems, rendering them ineffective (
31). Mutations of repressor genes can lead to overexpression of antibiotic-inactivating enzymes, such as the
β-lactamase
AmpC, leading to third-generation cephalosporin resistance (
32).
Horizontal gene transfer is ubiquitous in bacterial communities and represents the major mechanism by which bacteria are able to adapt to hostile natural environments and to antimicrobial innovation. Bacteria may exchange DNA information through transformation (incorporation of exogenous DNA from the surrounding environment), transduction (transfer of genetic material via bacteriophage vectors), or conjugation (direct cell-to-cell transfer of genetic material) (
26). Horizontal transfer of resistance genes is facilitated by their location on mobile genetic elements, such as integrons, transposons, and plasmids. The permutations of mobile elements are vast: integrons are found alone, or inside transposons; both integrons and transposons can be carried by plasmids or bacteriophages (
33). Such variations allow for gene shuffling and provide bacteria with vast capacity to adapt to changing environments or antimicrobial pressure, particularly in situations where large numbers of bacteria live in communities (such as the gastrointestinal tract or environmental reservoirs) (
34).
The rapid spread of extended spectrum
β-lactamases among Enterobacteriaceae world-wide has been a closely studied model of horizontal gene transfer. For example, the CTXM extended-spectrum
β-lactamase gene (
blaCTX-M) is thought to have mobilized multiple times from chromosomal DNA of
Kluyvera into plasmids, which have subsequently spread through conjugation into other Enterobacteriaceae species; such events have happened worldwide (
35,
36). Unfortunately, mobile elements frequently carry multiple resistance mutations simultaneously, allowing for broad resistance to be conferred quickly from one bacterial species to another. Plasmids that carry the carbapenemase gene
blaNDM-1 have also been shown to carry aminoglycoside, macrolide, rifampin, and sulfamethoxazole resistance genes, creating Enterobacteriaceae that are nearly pan-resistant (
37).
OVERVIEW OF SPECIFIC RESISTANCES
Several sobering trends in resistance have been observed over the years (
Figure 15.1,
Table 15.3). A surge in aminoglycoside resistance became a chief concern in the 1970s and 1980s, particularly in healthcare-associated Enterobacteriaceae and
P. aeruginosa. Although aminoglycoside importance and use has since declined with the development of alternative and often safer antimicrobials, such as advanced-spectrum
β-lactam antibiotics and fluoroquinolones, aminoglycoside resistance rates have been stubbornly persistent (
38). Rates of tobramycin resistance among Enterobacteriaceae sampled in the United States increased from 1.7% to 8.8% from 1999 to 2008 (
39). Aminoglycoside resistance is probably propelled by co-selection with other antibiotic resistance genes, such as fluoroquinolones (
40).
The availability of second-generation cephalosporins (such as cefoxitin and cefuroxime), third-generation cephalosporins (such as ceftriaxone and ceftazidime), and of
β-lactam-
β-lactamase inhibitor combination agents (such as piperacillin-tazobactam) has highlighted an additional set of resistance risks in gram-negative bacilli. For instance,
Enterobacter spp. were initially considered susceptible to cephalosporins but frequently developed resistance during therapy. The culprit was a spontaneously derepressed intrinsic chromosomal
AmpC β-lactamase (
41). Further discovery of plasmid-mediated
β-lactamases (such as ESBLs) conferring broad resistance to various penicillins and cephalosporins has made many gram-negatives such as
E. coli and
Klebsiella spp. difficult to control without turning to “antibiotics of last resort” such as carbapenems. The earliest recognized ESBLs evolved by point mutations from common, older plasmid-borne enzymes and were primarily found among hospital-acquired gram-negative organisms, particularly
Klebsiella spp., from the early 1980s to the late 1990s. However, since 2000, the CTX-M family of
β-lactamases has become increasingly dominant, displacing other
β-lactamases and invading both community and hospital reservoirs through its association with
E. coli in addition to
Klebsiella spp. (
42).
Because carbapenems are currently the broadest-spectrum antimicrobials commercially available, and because they are critical in treating ESBL-producing bacteria as well as other highly resistant gram-negative organisms such as
A. baumannii, the increases seen in carbapenem resistance have been alarming. Carbapenem resistance is mediated by various mechanisms, such as loss of the outer-membrane proteins and upregulation of efflux systems (
43). In the past decade, many different plasmidmediated carbapenemases—broad-spectrum
β-lactamases that hydrolyze carbapenems as well as other
β-lactam antibiotics— have emerged and spread worldwide, raising the risk of untreatable infections (
37). The emergence of multidrug-resistant strains of gram-negative bacteria that produce New Delhi metallo-
β-lactamase (
NDM-1) carbapenemase, as well as the remarkable ability of the
NDM-1 gene to disseminate across gram-negative species including community-acquired bacteria such as
E. coli,
Shigella boydii, and
Vibrio cholerae, will present perhaps the biggest challenge to IC to date (
44,
45).
The prevalence of trimethoprim and sulfonamide resistance in gram-negative bacteria remains a concern primarily in outpatient settings, where oral trimethoprim alone or trimethoprim-sulfamethoxazole (TMP-SMX) combination is often prescribed empirically for the treatment of urinary tract infections (UTIs). Resistance to trimethoprim is mediated via alterations in the target enzyme dihidrofolate reductase (
46), while resistance to sulfonamides is mediated by alterations in its target enzyme dihydropteroate synthase (
47). These resistance
genes are often linked to other resistance genes on transmissible elements, allowing for efficient spread and indirect selective pressure from associated antibiotics. Over the past decades, there has been an increasing trend in TMP-SMX resistance over time; surveillance data from 2000 to 2010 among US urinary
E. coli isolates showed an increase from 17.9% to 24.2% (
48). Reductions in trimethoprim use alone do not appear to be sufficient to reduce the rate of trimethoprim-resistant
E. coli, probably explained by the low fitness cost of trimethoprim resistance and the co-selection of trimethoprim resistance when alternative antibiotics are used (
49,
50).
Methicillin resistance has been a major concern in staphylococci since the 1980s. In 63 U.S. hospitals from 1974 to 1981, the percentage of
S. aureus infections resistant to methicillin increased modestly from 2.4% to 5%, due primarily to epidemics in four large teaching institutions (
51). In 1992, the pooled percentage of resistance had risen dramatically to 32.1%, and in 2004, to 53% (
52,
53). Methicillin resistance in healthcare-associated
S. aureus and coagulase-negative staphylococci is endemic in most U.S. hospitals.
Fluoroquinolones target enzymes responsible for bacterial DNA replication, such as DNA gyrase and topoisomerase, giving them broad efficacy against many gram-negative, and some gram-positive, organisms (
54). Since the 1980s, their potency and oral bioavailability have made their use widespread in treatment of infections (particularly pulmonary, urinary, and gastrointestinal), as well as in prophylaxis (e.g., in neutropenic patients). Widespread increases in fluoroquinolone resistance have been reported in many bacteria, in particular Enterobacteriaceae,
P. aeruginosa,
Streptococcus pneumoniae, and
Staphylococcus. aureus (
55,
56,
57). Ciprofloxacin resistance among
E. coli urinary isolates from the United States increased from 3% to 17% from 2000 to 2010 (
48). Although mutations to fluoroquinolone targets and efflux mechanisms explain some of the increase in resistance rates, the discovery of two disseminated classes of plasmid-mediated fluoroquinolone resistance mechanisms (
Qnr proteins that interfere with fluoroquinolone interaction with DNA gyrase and fluoroquinolone-modifying enzymes (
aac(6′)-Ib-cr)) also accounts for the explosive worldwide increase in fluoroquinolone resistance over time (
58).


 Occupational Health Services
Occupational Health Services
 Epidemiology and Control of Healthcare-Acquired Infections in Limited-Resource Settings
Epidemiology and Control of Healthcare-Acquired Infections in Limited-Resource Settings
 The Intensive Care Unit, Part A: HAI Epidemiology, Risk Factors, Surveillance, Engineering and Administrative Infection Control Practices, and Impact
The Intensive Care Unit, Part A: HAI Epidemiology, Risk Factors, Surveillance, Engineering and Administrative Infection Control Practices, and Impact
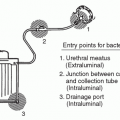 Urinary Tract Infections
Urinary Tract Infections
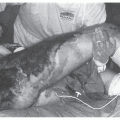 Infections of Burn Wounds
Infections of Burn Wounds



