Peter B. Ernst
Mucosal Immunity
Mucosal immune responses must discriminate between molecular signals that reflect a threat to the host versus those that are benign or even enhance health. This wisdom is achieved through mechanisms of antigen recognition, immune regulation, and the selection of effector responses that are tailored specifically to protect the delicate tissues and their physiologic functions. The adaptation of mucosal immune and inflammatory cells is shaped by literally millions of ligands in the local environment that are presented in an endocrine, paracrine, or autocrine manner. Although most of these ligands represent environmental antigens, they also include cytokines, growth factors, metabolites, hormones, and neurotransmitters. Moreover, the interaction between mucosal immunity and local organisms is recognized for its impact on the health of tissues well beyond the site where the original encounter occurs.
Specialization of Host Responses in Mucosal Tissues
The Challenge
Mucosal tissues not only provide a barrier between the host and the microbial world, but they are very delicate structures that perform important physiologic functions. For example, to maximize air exchange in the respiratory tract or nutrient absorption in the gut, these tissues have numerous morphologic adaptations that expand their surface area. It has been estimated that the surface area of the digestive tract is equivalent to one or two tennis courts, and this surface is exposed to myriad environmental antigens, whether they are toxins, food, or microbes. Further, humans alter their habits so these stimuli vary as we change our location, diet, and encounter new microbial challenges. Other realities of mucosal immunity include the challenge for host responses to reach luminal antigens “offshore” and protect against infection without compromising physiologic functions. Thus, immune and inflammatory cells in mucosal tissues adapt to changes in stimuli to acquire a pattern of differentiation that provides protection without disrupting adjacent tissues.
The gastrointestinal tract provides an informative model to illustrate the unique properties of mucosal immunity. In the gut, there are 10-fold more bacterial cells than host cells, and the microbiome entails several 100-fold more genes than the human genome. In addition, there are large numbers of viruses, fungi, and parasites. In response to this microbial burden, the gut has as many lymphocytes as a solid organ such as the spleen. Moreover, the gut secretes approximately 75% of the antibody produced every day. Other mucosal tissues share similar challenges and require many comparable adaptations in their local immune responses.
Adaptation of Mucosal Immune Responses
The concept of a “common mucosal immune system” emerged based on the related interface separating mucosal tissues from luminal stimuli. Further, the presence of dimeric immunoglobulin A (IgA) is an adaptation found in most mucosal secretions. Humans differ from mice in that they have the IgA1 and IgA2 isotypes that are represented to varying degrees in different sites. As described in Chapter 5, the structure of IgA reflects its adaption to the mucosal environment because it is actively transported across the epithelium via the polymeric immunoglobulin receptor (pIgR), resists proteolysis as well as acid hydrolysis, and serves to prevent the binding of pathogens or toxins without activating complement and its inflammatory effects. Paradoxically, IgA nephropathy is an IgA-mediated disease that is driven by the deposition of IgA1, which leads to the impairment of glomerular function.1 Thus, even with adapted systems, responses can go wrong and contribute to disease instead of protection.
After the discovery of IgA as the dominant immunoglobulin isotype in mucosal tissues, Ogra and colleagues2 demonstrated that oral immunization with a live-attenuated polio vaccine induced both IgG and IgA antibody responses in the serum and IgA in secretions, whereas systemic immunization with an inactivated virus had little effect on mucosal immunity. Evidence for the exchange of immunologic information among different mucosal tissues emerged from studies of mucosal immunization. When antigens are administered orally, for example, secretory IgA can be detected in other secretions such as tears. The detection of antibody in multiple tissues is largely attributable to the selective circulation of antigen-specific B cells from one mucosal tissue to another.3
Immunity includes a variety of responses, some of which neutralize the intended target but also mediate collateral damage to host tissues. In mucosal sites, the antigen burden requires a very delicate homeostatic response. A major contribution to homeostasis is the acquired tolerance to antigens that persist in the lumen of either the digestive tract (oral tolerance)4 or airway.5,6 This response allows the host to avoid adverse immune-mediated diseases resulting from environmental antigens and to “tolerate” the microbiota that persist in the lumen. The ability of local host responses to adapt to changes in luminal content, because of major changes in diet or microbial communities, exemplifies the flexibility in immune regulation that is needed to prevent overly exuberant responses to benign local antigens.
Although a multitude of factors shape mucosal immune responses, the selective accumulation of cells with a preferred phenotype for mucosal homeostasis occurs because of a limited array of biologic processes.7 The selection of a preferred phenotype in mucosal tissues can be achieved by deletion of cells with an unwanted phenotype, selective homing of cells with the preferred phenotype to the mucosae, differentiation of cells toward the preferred phenotype, and/or expansion of cells through proliferation (Fig. 7-1). With respect to proliferation, it should be pointed out that not all daughter cells are “identical twins.” Proliferation in lymphocytes is often asymmetrical, that is, the engagement of receptors in the immunologic synapse leads to the migration of receptors and their associated signaling molecules toward one pole. When cell division begins, the signaling molecules inherited by the daughter cells are asymmetrically distributed, and this necessarily changes the pattern of gene expression.8–10 This process may be relevant for changes in immune cell phenotype, particularly in association with disease.
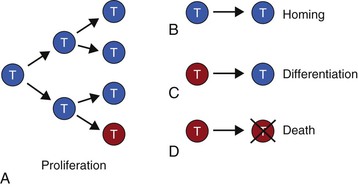
These four processes contribute to the phenotype of inflammatory cells, antigen-presenting cells (APCs) as well as T and B cells found in mucosal tissues. This understanding has been obtained by scientists accepting the challenge to study mucosal immune/inflammatory cells directly. One approach has used lavage of the airway or reproductive tracts as a window to view the lineage and function of cells in these sites. Although many investigators have studied mucosal cells directly in the gastrointestinal tract, because of technical challenges, cells such as neutrophils are rarely isolated from these tissues, whereas cells in the urogenital tract, salivary glands, and eye await more extensive characterization. As creative techniques to investigate these cells are developed, we can expect novel insights will emerge that will be important to consider in a comprehensive understanding of mucosal immunity.
Induction of Mucosal Immune Responses
Epithelial Cell Responses and Immunophysiology
Although single factors rarely explain complex biology, the interactions in mucosal immunity benefit from some insight into the individual elements. First, one should consider the interface between the host and its microbial milieu. In mucosal tissues, this is invariably the epithelial barrier, its mucous coating, and a variety of host factors that modify the microbial niche.11–15 Mucosal epithelial cells provide a physical barrier that limits the translocation of microbes or their metabolites into the underlying layers. The M cells are a specialized epithelial cell overlying the Peyer’s patches and serve as preferential sampling sites for the uptake of luminal antigens.16 Many microorganisms are preferentially taken up by the M cells.17,18 Similar secondary lymphoid structures can also be found in the nasal-associated lymphoid tissue of the airway. Of interest, cells resembling M cells are scattered throughout the epithelium of the gastrointestinal tract,19 suggesting that their enhanced ability to sample is more widespread than the Peyer’s patches.
Epithelial cells can express receptors for organisms such as human immunodeficiency virus (HIV),20 respiratory syncytial virus (RSV),21 Helicobacter pylori22–24 or Salmonella spp.,25 to name a few. These interactions favor colonization as well as invasion, which together lead to signaling events and the induction of mucosal immune responses.12 Epithelial cells in the airway,26–28 digestive tract,29–31 and urogenital tract32,33 respond to infection, toxins, and inflammation with the release of cytokines that not only alert the host of potential damage or danger but focus the responses to the site of injury based on a gradient of chemokines and other factors.34 Thus, epithelial cells are able to sense a danger signal and transduce responses that recruit and activate other immune or inflammatory cells.
Some epithelial cells are highly specialized to produce antimicrobial factors or express the pIgR to mediate the transfer of polymeric IgA into the lumen. The ability to transport IgA is shared by most epithelial cells and increased during inflammation. However, the secretion of antimicrobial factors tends to be more localized. For example, saliva contains secretory leukocyte protease inhibitor,35 gastric parietal cells produce acid, and other epithelial cells produce a range of factors, including lysozyme, lactoferrin, defensins,13 and antibacterial lectins11,36 that interfere directly with microbial growth or modify physiologic function to protect the host. One example of altered physiology in host defense is the role played by secretions to create a pressure gradient that impairs colonization in the deep regions of mucosal glands. Further, highly adapted processes, such as the mucociliary apparatus in the lungs, aid in the elimination of environmental contaminants and microbes.
In the gut, motility partners with the secretions to create a catharsis that contributes to the clearance of infections. This concept was illustrated in studies of immunity to nematodes. These organisms induce a robust Th2 response associated with the accumulation of IgE and mucosal mast cells.37 Degranulation of mast cells releases mediators that contribute to epithelial cell secretion and an increase in motility that, when impaired, decreases the clearance of several nematode species.38–40 These physiologic responses are regulated by neuroendocrine cells, often triggered by microbial products41,42 as well as cytokine responses that contribute collectively to mucosal immunity because they facilitate the clearance of infections. Neuroendocrine factors also regulate immune cell function. In short, the protective or pathogenic effects of mucosal immune and inflammatory cells cannot be separated from the other cells and mediators within the tissue. This raises the possibility that drugs impairing motility may not always be helpful in the control of diarrhea.
Antigen-Presenting Cells
The mucosal tissues have a full complement of macrophages and dendritic cells. These cells are found throughout the lamina propria, although some are enriched in secondary lymphoid structures, including Peyer’s patches in the gut, similar subepithelial aggregates in the airway or other tissues, as well as in the draining lymph nodes. It has been reported that some dendritic cells extend their dendrites into the lumen and sample antigens directly.43,44 In other studies, phagocytes have been shown to engulf apoptotic epithelial cells45,46,47 that, if infected, could carry microbes and create another mechanism of antigen sampling.48,49
It is increasingly clear that various factors within the mucosa select for unique APCs50 that facilitate the accumulation of helper T (Th) cells that favor the induction of tolerance and/or mucosal IgA. Thus, there is a feed-forward as cells involved in the recognition and processing of antigen acquire a mucosal phenotype that, in turn, favors the expansion of Th cells that select for effector mechanisms, such as IgA or mucosal mast cells that culminate in an ideal mucosal immune response. For example, initial engagement of nucleotide oligomerization domain 2 (NOD2) expressed by APCs, with muramyl dipeptide (MDP) administered intranasally, stimulates the production of thymic stromal lymphopoietin (TSLP) and the induction of Th2 cells in the lung.51 Similarly, initial stimulation of NOD2 in the gut or in the context of ocular toxoplasmosis52 induces proinflammatory responses. However, repeated stimulation with NOD2 ligands renders APCs nonresponsive to ligands for NOD2 or Toll-like receptors (TLRs),53,54 as evidenced by decreased production of interleukin-1β (IL-1β), IL-8, and tumor necrosis factor-α (TNF-α).54 Mutations in the NOD2 gene are associated with Crohn’s disease,55 possibly by impairing the ability of APCs to adapt to these repeated signals from microbial ligands.
Innate Lymphoid Cells
Complementing the transition from innate to adaptive responses is a population of innate lymphoid cells (ILCs). These cells are non-B, non-T cells with a lymphoid morphology.56,57 Historically, ILCs included natural killer (NK) cells as well as the lymphoid tissue inducer (LTi) cells. Now it is clear that there are other subsets that are closely related to most, if not all, Th-cell subsets because they express similar transcription factors and patterns of cytokine production. To clarify this area, a new nomenclature has been proposed, in which ILCs are divided into subsets referred to as ILC1, ILC2, and ILC3.57 Of interest, ILC1 and ILC2 express transcription factors and cytokines that resemble Th1 and Th2 cells, respectively, whereas ILC3 include cells resembling Th17 or Th22 cells, as well as LTi cells (Fig. 7-2),57 thus giving a functional homologue of Th-cell subsets to innate immunity.
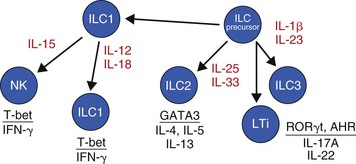
ILCs reside in the tissue constitutively and become activated by some of the cytokines released by APCs, epithelial cells, and T cells. In turn, ILCs express the transcription factors and cytokines that are appropriate to amplify the desired host response for the current antigenic signal. Given that adaptive immunity takes several days to be induced, ILCs complement the role played by macrophages and other innate cells in limiting infections as well as enhancing the milieu to select for the appropriate T-cell and B-cell responses that eventually confer adaptive immunity and memory.
Further evidence of the integration between ILCs and Th cells is found in the response of ILCs to Th-derived cytokines. For example, IL-17A or IL-22 from Th17 cells has been reported to activate NK cells and enhance immunity to fungi in mucosal sites such as the kidney58 and oral cavity.59 Other Th-cell cytokines activate cells involved in acute/innate inflammatory response in different ways. For example, Th2 cells enhance the accumulation of mast cells, basophils, and eosinophils, as well as increase IgE antibodies that enable IgE-binding cells (such as mast cells and basophils) to recognize antigen and release their mediators that contribute to clearance of nematodes.37
Sensing Mucosal Microbiota
The importance of the microbiota in regulating host responses begins with the microbes having access to the host and the subsequent recognition of their molecular structures.60–62 The sensing of microorganisms is mediated through pathogen-associated molecular patterns (PAMPs).63 The PAMPs are detected by pattern recognition receptors (PRR) expressed on the surface of several cell lineages. There are several classes of PRR, including TLRs63; NOD-like receptors (NLR), including the NOD family64; Rig-like helicases (RLH)65; and various proteins containing thrombospondin repeats, such as thrombospondin 1,66 mindin,67 and the recently described brain angiogenesis inhibitor 1 (BAI1).25 In addition to bacterial factors, tissue damage, including debris from cells dying of apoptosis or necrosis, provides an array of molecules referred to as alarmins or danger-associated molecular patterns (DAMPs).68 Further, the release of adenosine triphosphate (ATP) from dead cells in damaged tissue is important for additional phagocyte recruitment69,70 and the regulation of inflammation that contributes to tissue repair.
Many PRR engage their ligand on the cell surface, whereas others are activated in phagosomes or elsewhere in the intracellular space after translocation of the PAMPs. Soluble PAMPs in the blood, such as endotoxin, can activate surface receptors directly, although concentrations of these ligands rarely reach the thresholds needed to activate leukocytes ex vivo. Microbes, exemplified by H. pylori, remain primarily on the cell surface of (gastric) epithelial cells, where surface PRR may be engaged. Additional activation occurs subsequent to the translocation of bacterial effector molecules into the cytoplasm, where peptidoglycan can induce IL-8 responses after sensing by NOD1.71 Other organisms, such as enteroinvasive Escherichia coli or various species of Salmonella, enter a cell by means of invasion as well as engulfment.72 The internalization process creates phagosomes that can juxtapose the PAMPs with PRR within the phagosome, where they activate their respective responses.
Subsequent to the engagement of PRR, signaling pathways are stimulated, leading to the activation of transcription factors and the production of various host response molecules. Perhaps the best-studied is the role of the myeloid differentiation primary response protein 88 (MyD88) as an integral part of the signaling for several of the TLRs.73,74 The fact that several PRR share a signaling pathway creates a synergy that may augment the potential stimulation from relatively small concentrations of PAMPs. Accumulating PAMPs within a phagosome would further enhance their concentration and potential for signaling.
Several distinct host responses are induced after sensing by PRR. One of the first would be the production of chemokines that recruit and activate other immune cells to the site of damage. Other responses include changes in the expression of accessory molecules on APCs that contribute to T-cell activation, oxidative burst, and the production of reactive nitrogen species, various vasoactive mediators, or arachidonic acid metabolites.
An important function of innate cells is to clear microbes or damaged tissue by phagocytosis. Engulfment of sterile debris or benign microbes leads primarily to the production of anti-inflammatory cytokines such as IL-10 or transforming growth factor-β1 (TGF-β1).75,76 In contrast, clearance of pathogens usually leads to the production of cytokines that activate various Th-cell subsets. Another consequence of internalization of antigen into dendritic cells is its presentation to CD4 or CD8+ T cells, leading to T-cell activation. It should be pointed out that epithelial cells often express molecules associated with T-cell activation, including those recognized by invariant natural killer T (NKT) cells77 but also T cells.78,79 An interesting nonmicrobial example is the rapid uptake of gliadin by duodenal epithelial cells.80 Subsequent to this uptake, gliadin is processed by tissue transglutaminase to create a peptide that fits well into the DQ2 or DQ8 peptide-binding pocket and leads to Th-cell activation and celiac disease.81 Microbial antigen uptake and processing by epithelial cells or APCs by similar or distinct mechanisms would enhance adaptive immunity to an organism.
Sensing Metabolites
The PAMPs and DAMPs recognized by the host represent a fraction of the possible signals that shape mucosal immune responses. The broad collection of microbial and host metabolites constitutes a much larger pool of ligands that is just beginning to be appreciated. The value in understanding metabolomics can be illustrated with a few examples.
Several white cell lineages, including dendritic cells and Th cells, express a receptor that binds different ligands, including polycyclic aromatic hydrocarbons, suggesting environmental toxins modulate host responses.82 The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that regulates the expression of many immune genes. In addition to toxins, natural ligands can be generated from dietary tryptophan by the initial enzymatic activity of indoleamine 2,3-deoxygenase (IDO) to yield kynurenine. Kynurenine can accumulate in the placenta and induce regulatory helper T (Treg) cells that may help prevent rejection of the semiallogenic fetus.83 Further, catabolism of dietary tryptophan in the gut by bacteria or fungi to other AHR ligands can assist in the induction of IL-22, which binds to epithelial cells and stimulates the release of antimicrobial peptides.84,85 Thus, microbiota and the host compete for the same substrates to generate different metabolic products that may modify mucosal immune responses toward Treg or Th22 cells85 while other AHR ligands can select for Th17 cells.86
Purine metabolism represents another pathway in which bacteria and the host compete for the same substrate.87 In brief, the accumulation of ATP from bacteria88 or from dying cells69 can be metabolized to adenosine diphosphate (ADP), 5′-adenosine monophosphate (5′-AMP), and then to adenosine.87,89 ATP can be a proinflammatory signal that favors the accumulation of Th17 cells in the mucosa,90 whereas the accumulation of adenosine depletes this stimulus and usually confers an anti-inflammatory activity mediated by the A2A adenosine receptor (A2AAR).87 Although Treg cells can generate adenosine as a mediator of suppression, bacteria can also achieve this to suppress innate responses to favor colonization.91
Another example of the role for metabolism in regulating host responses is the conversion of vitamin A to retinoic acid by dendritic cells expressing retinol dehydrogenase.92 This process plays an important role in conditioning leukocytes with a mucosal phenotype93,94 and imparting the ability for these cells to home to mucosal tissues.92,95 Further, APCs exposed to retinoic acid assume the capacity to direct Th-cell differentiation away from Th17 cells and toward Th2 and Treg cells96 that possess the ability to increase IgA production.97,98 One can only imagine how new data emerging from studies of the microbiome will identify other metabolic reactions that contribute to the molecular environment shaping mucosal immune response and the microbial niche.
Discrimination between a “Commensal” and a “Pathogen”
The crux of the matter for mucosal immune responses in health and disease is being able to know when and how to respond. For years, microbes have been described as being “commensals” or “pathogens;” however, these categories represent opposite ends of a spectrum that defines the interrelationship between the host and its microbial communities. As discussed later, this is illustrated by organisms that can exist as commensals, but in the context of the host response, they can assume a biologic role that is decidedly disadvantageous to the host.99
The conundrum of distinguishing pathogens from commensals emerges because the latter can also express ligands for PRR yet do not impart a strong danger signal.53 There are several reasons to explain this paradox. It may be related to the burden of a particular microbial species, the age at which infection occurs or its duration, the structure of its PAMPs that may render them less proinflammatory, or possibly the absence of secretion systems, invasion mechanisms, or other virulence factors. At the other extreme, one encounters pathogens that display an array of molecular patterns that allows them to send a different, perhaps “fresh” or more intense set of signals that stimulates mucosal responses immediately.
Microbes produce hundreds of metabolites and molecules, creating a cascade of stimuli that begins with binding to host epithelial cells; sometimes using secretion systems that translocate bacterial effector molecules into host cells; invasion and/or internalization; engagement of numerous extracellular or intracellular pattern recognition receptors (such as TLRs, NODs, etc.); and the initiation of chemokine and cytokine production. As described by Matzinger,100 this process signals “danger,” and the chemokines recruit and activate innate cells to phagocytose the offending microbe, amplify the host response, and induce appropriate effector mechanisms.100 This is illustrated by organisms such as Salmonella spp. that stimulate APCs to produce proinflammatory cytokines, including IL-6, IL-12, or IL-23, that stimulate the differentiation of Th1 or Th17 cells.101,102 Although these cells contribute to protective functions, host responses must be regulated carefully to avoid immune-mediated damage and to maintain the physiologic functions that are required to sustain life.
Contrasting the inflammatory responses leading to a relative sterile immunity are infections that induce a degree of tolerance. For example, dendritic cells exposed to H. pylori favor the ability of these APCs to induce Treg cells103,104 and contribute to persistent infection. Indeed, a pathogen such as H. pylori can coexist more or less peacefully for the host’s entire life.46,47 The molecular basis to explain this effect of H. pylori remains unclear but could include the modest inflammatory effect of its lipopolysaccharide (LPS)105 or any number of metabolic factors that remain to be defined. Adding to the complexity of this biology, H. pylori may even confer some advantages to health in the digestive tract106 or other sites as discussed later. Of importance, “sterile immunity” may be less advantageous to the host than a more mutually beneficial relationship.
Adaptive Immunity to Mucosal Stimulation
The transition from innate to adaptive immunity is based on the interaction among Th cells, APCs, and the molecular environment that drives Th-cell differentiation. As illustrated in Figure 7-3, the cytokine milieu induced after antigenic stimulation shapes the differentiation of Th cells. The combination of microbial factors, the conditioned responses of APCs that have adapted to their respective niche, and local cytokines (e.g., from ILCs or other T cells), all regulate Th-cell differentiation. This is illustrated with nematode infections that induce TSLP and IL-4, leading to the differentiation of naïve Th cells into Th2 cells.107–109
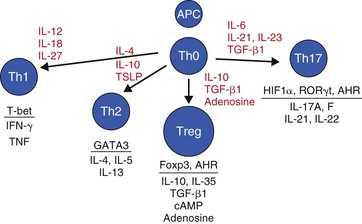
Th17 cells provide another important Th-cell response in mucosal tissues. IL-17 production was first implicated in mucosal infections in studies of the pathogenesis of H. pylori in human gastric tissue.110 Subsequently, Th17 cells, through the production of IL-17 and IL-22, have been shown to play an important role in immunity to mucosal pathogens,56 including Citrobacter rodentium,111 Klebsiella pneumoniae,112 H. pylori,113 and Candida albicans.59
Most inflammatory responses are mixed because cytokines capable of favoring different Th-cell subsets are induced simultaneously. The plasticity of Th cells and the complexity of Th-cell differentiation are attributable to the fact that multiple cytokines targeting a Th cell induce the same, complementary, or competitive signaling pathways. Although some responses can be uniform, for example, Th2 responses induced by nematodes114 or Th1 responses induced by Mycobacterium tuberculosis,115 other microbes induce mixed Th1/Th17 responses, and there are even reports of Th1 responses in Treg cells.116
The Th cell–derived cytokines expand and differentiate CD8+ cytotoxic T cells (CTL) or B cells. In addition, these cytokines feedback on innate cells to increase the expression of cytokines produced by epithelial cells, ILCs, or APCs. For example, the production of interferon-γ (IFN-γ), IL-2, and IL-15 are particularly effective at increasing cytotoxic activity mediated by NK cells, invariant NKT (iNKT), and CTL that would be effective at mediating immunity against intracellular pathogens, including viruses and some invasive bacteria. These responses would be induced most efficiently by Th1 cells in an environment in which the pathogen or vaccine antigen induced IL-1 and IL-12, for example.
Mucosal B cells are noted for undergoing isotype switching from IgM-bearing cells to IgA-producing cells. Although IgA is usually the predominant antibody in mucosal tissues, other isotypes can be induced, including various isotypes of IgG and IgE. IgG tends to be the predominant antibody isotype in the lower airway and reproductive tract. The process of isotype switching is mediated by different cytokines, including IL-4, which favors IgE117 and IgG1, as well as TGF-β1, B-cell–activating factor of the TNF family (BAFF), or A-proliferation–inducing ligand (APRIL), which direct switching to IgA.118,119,120 Of interest, one of the richest sources of TGF-β1 is the Treg-cell subset.98 Treg cells induce more IgA-producing cells than Th2 cells, in addition to inhibiting proinflammatory cytokine responses. This apparent paradox illustrates the delicate balance that is required in mucosal immune homeostasis. Inhibition of host responses by Treg cells can favor persistent infection and the development of commensalism. Commensals may not be cleared by IgA alone, or their profile of antigens and virulence factors may not favor the induction of sufficiently strong cognate B-cell or other host responses that would eradicate the infection. However, other microbes induce host responses that eventually eliminate and often prevent future infection. Treg cells may help select for IgA antibody production that is sufficient to protect the host while simultaneously limiting tissue damage that could emerge from excessive inflammation that might be induced by pathobionts.98 Subsequent to the isotype switch to IgA, other Th-cell–derived cytokines (e.g., IL-5, IL-6, and IL-10) can expand the pool of IgA-producing cells121 and coordinate an effective host response that remains under the control of Treg cells.
The production of IgE in the airway and digestive tract occurs in the context of nematode infections or allergies. Environmental triggers, including microbial PAMPs, can stimulate the production of cytokines that favor Th2 differentiation and IgE production.51 Further, rhinoviruses have also been implicated as a trigger that exacerbates atopy, including food allergy and wheezing.122 Thus, careful regulation of mucosal immune responses is important for maintaining immunologic homeostasis to infections.
Immunologic Homeostasis in Mucosal Tissues
Immune responses in mucosal tissues, including the gut, airway, urogenital tract, and eye, are regulated by CD4+ Th cells, regulatory NKT cells, CD8+ T cells, and B cells. In response to benign antigens, which on their own do not deliver a loud danger signal, immune cells with a “regulatory” phenotype maintain immunologic homeostasis. Lineages with this function are represented unevenly in different tissues, but together they create a regulatory network to maintain equilibrium in mucosal immune reactivity.
Helper T cells are an important part of the adaptive immune system that contributes to the balance of immunity and immunologic restraint required for healthy mucosal tissues. A number of functionally distinct CD4+ Th-cell subsets have been elucidated, including Treg cells, which mediate anti-inflammatory activity. These cells are a topic of intense study because of their ability to control inflammatory responses and prevent autoimmune reactions.
Treg, or various Th subsets with regulatory function, inhibit immune and inflammatory responses through the production of IL-10 and/or TGF-β1. In addition to these cytokines, interactions between surface cytotoxic T-lymphocyte antigen-4 (CTLA-4) on Treg cells and receptor molecules on target cells limit immune responsiveness. Other data suggest that contact between Treg and effector T (Teff) cells stimulates the production of the anti-inflammatory mediator IL-35.123 A more intimate mechanism of intercellular contact was described by Bopp and co-workers,124 who reported that the suppressive action of murine Treg cells depended upon the transfer of cyclic adenosine monophosphate (cAMP) from Treg to responder T cells. Although pharmacologic manipulation may increase the levels of cAMP available to mediate Treg-cell function, there are biologically relevant stimuli that accomplish this. One example is adenosine.
There is now substantial evidence that adenosine is an important mediator of Treg-cell function. For example, engagement of the A2AAR not only induces Treg cells125 but is also required for optimal Treg-cell function.126,127 Subsequently, investigators reported that Treg cells had the ability to synthesize adenosine through the action of two ectoenzymes: CD39 (ectonucleoside triphosphate diphosphohydrolase-1 [NTPDase-1]) and CD73 (ecto-5′-nucleotidase). Many Treg cells in mucosal tissues express CD39 and CD73, which catalyze the transformation of ATP to adenosine.128 ATP enhances dendritic cell maturation129 and increases Th17 responses in the intestinal lamina propria.90 Because most Treg cells express CD39 and CD73, they not only generate adenosine, which inhibits many responses, including Th17 cells,130 but degrade ATP and, in so doing, remove this proinflammatory stimulus from the immunologic synapse. As described earlier, ATP metabolism provides an intersection between the host and microbes that generate and use these products for their respective advantage.
The induction of regulatory cells to infections has also been examined in the airway.131,132 For example, infection with Bordetella pertussis stimulates the accumulation of Treg cells that can attenuate injury caused by immune and inflammatory cells induced by infection.133 Braat, Mills, and co-workers134 have expanded this model to identify that the parenteral administration of an immunomodulatory antigen from Bordetella pertussis protects against colitis induced by the adoptive transfer of CD45RBhi Th cells. This experiment shows that induction of one functional subset of Th cells by a specific microbial antigen can create a regulatory network that interferes with inflammation mediated by Th cells, even in other mucosal tissues. This notion is supported by the observations that Th2 responses induced by nematode infections attenuate disease mediated by Th1 and/or Th17 cells in the stomach135 or intestine.136,137 These reports provide evidence that immune deviation induced by mucosal infection deflects a potentially adverse host response to a more tolerable condition. Of importance, these studies also demonstrate that new knowledge gleaned from research in one tissue or with one organism may have relevant applications in the management of other diseases in unrelated tissues.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


