Identifying Appropriate Doses and Schedules of Molecularly Targeted Agents
In the early years of targeted therapy development, there was enthusiasm for the concept of determining the “optimal biological dose” (OBD) for targeted agents rather than their maximum tolerated dose (MTD).
123 The OBD concept was based on the premise that the narrow therapeutic window for conventional cytotoxic agents would not apply to targeted agents, which because of their selectivity were hypothesized to show much greater differences between doses with biological and clinical activity and doses that produced unacceptable toxicity. However, experience with small molecule inhibitors developed as targeted agents has shown that most have narrow therapeutic windows, when a therapeutic window exists at all.
123 Examples of adverse events that limit dosing of targeted agents include rashes for EGFR inhibitors and multitargeted kinases,
124 stomatitis for mTOR inhibitors,
125 hypertension for VEGF pathway inhibitors,
126 diarrhea for Notch pathway inhibitors,
127,128 and vascular occlusion for the kinase inhibitor ponatinib.
129 Furthermore, since many small molecule targeted agents require continuous exposure for maximum effect, low-grade toxicities (e.g., rash or stomatitis) that may be tolerable when treatment is intermittent become intolerable when both treatment and toxicity are chronic. In more than 60% of cases, Phase 1 trials of small molecule targeted agents have had dosing limited by unacceptable adverse events.
130,131 Monoclonal antibodies differ from small molecule inhibitors in that their dosing is most commonly not limited by toxicity, but rather by reaching a dose that saturates their targeted binding sites.
123Successful use of a molecularly targeted agent in the clinical setting implies that the activity of the target has been successfully inhibited or blocked by the agent, with the depth of inhibition and the duration of inhibition being sufficient to achieve the desired effect. The depth and duration of inhibition required for antitumor effect can be empirically determined in preclinical in vivo models in which pharmacodynamic measures of target inhibition are measured at selected time points following agent administration and correlated to antitumor activity. Pharmacokinetic correlates of antitumor activity can also be determined, so that clinical development of the agent can proceed with both pharmacodynamic and pharmacokinetic benchmarks that need to be met in early phase clinical trials for the agent.
132 As multiple specimens for pharmacokinetic data are easier to obtain than multiple specimens for pharmacodynamic studies, it is beneficial to have robust preclinical data defining the pharmacokinetic exposures associated with anticancer activity so that these exposures can be targeted in the initial clinical trials of targeted agents.
Practical and logistical issues complicate the approach of basing dosing decisions on target effects within tumor tissue. Sequential tumor biopsies are needed to assess target modulation, with pretherapy and posttherapy sampling required. Intratumoral heterogeneity may produce differences between pre- and posttreatment tissues unrelated to agent effects, a factor that is exacerbated by the small size of tissue specimens often obtained with sequential biopsies.
133 Variability may also be introduced by the postexcision processing and storage of biopsy specimens, as changes in the status of signaling pathways in tumor specimens can occur quickly following loss of blood supply.
133 A final source of variability is that associated with the assay procedures themselves. As with any clinical laboratory procedure, obtaining interpretable data requires that issues such as assay sensitivity, specificity, precision, accuracy, and linearity be satisfactorily addressed. It is essential that laboratory methods be adequately studied and validated in preclinical models prior to their application in clinical trials to inform dosing decisions.
133Agent effects on surrogate tissues or on blood components may be used to inform dosing decisions. Some on-target effects on surrogate tissues may be clinically evaluable, such as the skin pigmentation changes caused by KIT inhibitors, the characteristic rash associated with EGFR inhibitors, and the rapid decrease in platelets that follows treatment with the BCL-2 inhibitor navitoclax.
134,135,136 Examples of changes in plasma proteins include the increase in VEGF that occur following treatment with small molecule inhibitors of VEGFR2 and the compensatory increase in IGF-1 and human growth hormone levels that follows administration of antibodies directed against the IGF-1R.
137,138 Differences between tumor and surrogate tissue in both extrinsic factors (e.g., degree of vascularization) and intrinsic cellular factors (e.g., the activity of drug efflux pumps and other resistance mechanisms) may limit the correlation between surrogate and tumor tissue molecular response to the targeted agent.
The complexities described above illustrate the challenges in performing phase 1 studies with dose escalation based on biologic endpoints determined by testing tumor tissue. As an example, while pharmacodynamic effects of the PARP inhibitor of veliparib could be documented at doses as low as 25 mg,
139 results of a phase 1 study showed that activity appeared greatest at the MTD (400 mg
twice daily) for patients with BRCA mutations.
140 An argument can be made for developing a targeted agent utilizing the highest dose with acceptable toxicity.
141 Under the assumption that the effect of a targeted agent may plateau (but will not decrease) with increasing dose, this approach ensures that further testing of the agent will be performed at a dose with maximum activity and acceptable toxicity.
141 However, even when dosing decisions are based primarily on toxicity or agent effect on surrogate tissue, it is important to define within one or more adult patient populations the effect of the agent on its target in tumor tissue at the dose and schedule being brought forward for further clinical evaluation. Documenting that the agent consistently achieves its desired target effect in tumor tissue for at least some cancers enhances confidence in the decision to go forward with further clinical development of the agent.
Precision Medicine and Clinical Trial Designs for Molecularly Targeted Agents
“Precision medicine” has been defined as “the tailoring of medical treatment to the individual characteristics of each patient.”
142 As noted in a National Academy of Sciences report, precision medicine “does not literally mean the creation of drugs or medical devices that are unique to a patient, but rather the ability to classify individuals into subpopulations that differ in their susceptibility to a particular disease, in the biology and/or prognosis of those diseases they may develop, or in their response to a specific treatment.” Inherent in this concept are two components: a treatment that provides specific benefit for a subpopulation of patients and a well-validated diagnostic test to reliably identify this group of patients. The latter component highlights the importance of the companion diagnostics that must accompany clinical development of molecularly targeted agents.
The U.S. FDA has articulated a policy that requires the coapproval of a diagnostic with a therapeutic product when the diagnostic is essential to the safe and effective use of the therapeutic product.
143 Examples of coapproval include trastuzumab and its companion diagnostic HercepTest
TM, vemurafenib and its companion diagnostic for BRAF V600 mutation, and crizotinib and its companion diagnostic using ALK break apart FISH probes.
143 These examples illustrate a “one drug, one test” pathway for coapproval of a therapeutic agent and its companion diagnostic, but this regulatory pathway will certainly yield to diagnostics that test for multiple biomarkers simultaneously. The application of “next generation sequencing” (NGS) to cancer specimens best illustrates this new pathway. While research applications of NGS have become widespread, the rigorous validation steps required for clinical application, including determining suitable analytical sensitivity, specificity, accuracy, and precision across the reportable range of genomic alterations, are more recent.
144 Capabilities now exist for using NGS methods to reliably characterize clinically relevant base substitutions, short insertions and deletions (indels), copy number alterations, and selected fusions in a single test across hundreds of cancer-related genes using routine formalin-fixed and paraffin-embedded (FFPE) clinical specimens.
144The ability to simultaneously determine for clinical use the status of hundreds of genomic alterations and the realization that only small subsets of patients will have any given genomic alteration has given rise to the development of new clinical trial designs for molecularly targeted anticancer agents. One example is the “basket” clinical trial design, which is a signal finding design in which eligibility criteria are agnostic to histology and only require the presence of a specific biological characteristic or genomic alteration.
145 The Imatinib Target Exploration Consortium Study B2225 was an early example of this design, as it allowed enrollment of patients with any malignancy with evidence for a potential role of one or more imatinib-sensitive tyrosine kinases.
25 Forty different malignancies were enrolled, with objective responses observed in five diagnoses with known genomic mechanisms of activation of imatinib target kinases. The initial pediatric clinical trial evaluating crizotinib (NCT00939770) used a basket-like approach for its phase 2 expansion, as patients with known genomic activation of ALK were eligible regardless of histology.
146 The pediatric phase 1-2 evaluation of vemurafenib used a similar design, with enrollment limited to patients with BRAF V600 mutation without regard for histology.
147The “umbrella” clinical trial design enrolls patients and then assigns them to one of multiple treatments based on the results of biomarker evaluations.
148 An example is the Lung Cancer Master Protocol (Lung-MAP, S1400, NCT02154490) for patients with advanced squamous cell carcinoma whose tumors have progressed after frontline therapy.
148 Patients have genomic analysis performed at study entry and are then randomly assigned to either an agent targeting the abnormal pathway or to standard second-line therapy as follows: PI3KCA mutations (PI3K inhibitor GDC-0032), alterations in CDK4/6, CCND1, CCND2, and CCND3 (CDK4/6 inhibitor palbociclib), alterations in FGFR1, FGFR2, and FGFR3 (FGFR inhibitor AZD4547), and alterations in HGF/c-MET (anti-HGF monoclonal antibody rilotumumab). Patients without an abnormality in one of these pathways are randomly assigned to either an immunotherapeutic approach using antiprogrammed cell death 1 ligand 1 (PD-L1) or standard chemotherapy (docetaxel or gemcitabine). A phase 2-3 design is used for each of the randomizations, such that if encouraging activity is observed in the initial phase 2 component for any biomarker/agent group, accrual can be extended to 300 to 400 patients for a definitive assessment of the agent for this genomically defined patient population.
The NCI MATCH clinical trial uses an “umbrella” design with multiple molecularly based phase II studies embedded within the overall trial.
148 Adult cancer patients with any solid tumor or lymphoma that has progressed after at least one standard therapy for metastatic or advanced disease are eligible and must undergo biopsy at disease recurrence to have their tumors characterized for actionable genomic alterations. Patients with a genomic alteration that matches one of the 20 to 25 study agents are assigned to the treatment arm for this agent. Agent/gene pairs are selected on the basis of data showing the targeted agent has demonstrated activity in a human tumor carrying the genomic abnormality. Agents known to be inactive against a certain histology (e.g., BRAF inhibitors targeting V600E in colon cancer) are not evaluated against that histology. The protocol prescribes rules for assigning patients to agents based on the molecular findings from their biopsy, such that a tumor board is not required for decision making. Target accrual for each treatment arm is approximately 30 evaluable patients, with all meeting the molecular eligibility criteria but having a mixture of malignancies.
The ALCHEMIST trial is focused on the adjuvant treatment of early-stage adenocarcinoma of the lung.
148 Among this patient population, approximately 15% have EGFR mutation and 5% have ALK gene fusion. Patients are screened for these genomic alterations, and those with EGFR mutation and randomized to receive erlotinib or not following standard adjuvant chemotherapy, while those with ALK gene fusions are similarly randomized to receive crizotinib or not following standard adjuvant chemotherapy. The primary outcome measure for each randomization is overall survival. To obtain the required number of patients for each randomization (approximately 400 patients per randomization), 8000 patients will need to be screened.
The ALCHEMIST trial highlights the challenges that face application of the precision medicine concept in the childhood cancer setting. The proportion of patients with adenocarcinoma of the lung that have EGFR mutations is similar to the proportion of neuroblastoma patients that have activating ALK mutations. While it is feasible to screen 8000 patients with lung cancer to identify the approximately 400 with EGFR mutation required to conduct a randomized phase 3 trial, to attempt the same for children with high-risk neuroblastoma and ALK mutations would require decades of accrual. Identifying activity signals of single agent activity is not the problem, since 20 or fewer patients are required
to convincingly document the ability of an agent to induce objective responses in a biomarker-defined patient population. Standard pediatric phase 2 trial designs include 20 to 25 patients,
149 and the activity of crizotinib was clearly documented by fewer than 10 patients enrolled into an expansion cohort of a phase 1 study.
146 The challenge is identifying the contribution of a targeted agent when it is added to standard therapy for a small, genomically defined patient population. The standard therapy has some level of effectiveness, and the traditional approach of conducting a randomized phase III trial to reliably identify the contribution that the agent makes when added to standard therapy is not feasible because of patient numbers.
The development of imatinib for Ph
+ ALL in children illustrates one approach to addressing this challenge.
150 In this case, results from a single-arm study convincingly demonstrated that imatinib added benefit to standard therapy. Three factors related to this example are noteworthy. First, imatinib had substantial single agent activity against the target patient population, increasing confidence that any effects observed with its addition to standard therapy were likely to be true effects. Second, there was a reasonably large, recent historical control population that allowed a comparison to be made between outcome for standard therapy with and without the addition of imatinib. Finally, the treatment effect observed with the addition of imatinib was very large, with 3-year EFS of 80% ± 11% with the addition of imatinib, more than twice that of historical controls (35% ± 4%).
150 The extent to which the imatinib example can be replicated for other agent/biomarker combinations will depend in part on the extent to which these factors are similarly represented. An example of applying the historical control approach to identify the contribution of novel agents added to standard therapy is the ANHL12P1 (NCT01979536), a randomized phase 2 clinical trial for children with anaplastic large cell lymphoma. This population is characterized by their genomic lesion (an ALK fusion gene) and by their uniform expression of the surface protein CD30, making these patients responsive to both crizotinib and brentuximab vedotin.
146,151 Patients enrolled on ANHL12P1 receive standard therapy plus either crizotinib or brentuximab vedotin, and a total of approximately 140 patients are to be enrolled. The primary outcome measure is a comparison of the EFS for each arm to the estimated EFS for chemotherapy alone, such that with 70 or fewer patients per arm, one or both arms may be identified as superior to chemotherapy alone.
An alternative clinical trial design for small patient populations is to randomize and target large treatment effects, with or without inflated Type I error. When large treatment effects are targeted, small patient numbers are required, as illustrated by the evaluation of a scorpion antivenom in children using a randomized design with a minimum sample size of 14 patients.
152 Likewise, using type I error rates greater than the standard two-sided 0.05 reduces the numbers of patients required for any given targeted effect size.
153 Because historical controls are often not available for genomically defined patient populations, randomization may be essential for identifying the contribution of a targeted agent when it is added to standard therapy.
Examples of applications of the precision medicine concept to pediatric oncology, in addition to those described above, include a clinical trial of the MEK inhibitor selumetinib for patients with low-grade astrocytoma (NCT01089101) and clinical trials of the Smoothened (SMO) inhibitors vismodegib and sonidegib for children with sonic hedgehog (SHH) pathway activated medulloblastoma (NCT01239316 and NCT01708174, respectively). In the former, a phase 2 expansion cohort is evaluating the activity of selumetinib for different genomically defined groups, including patients with the BRAF V600E mutation and/or KIAA1549-BRAF fusion and patients with neurofibromatosis 1 (NF-1). The evaluations of Hedgehog pathway inhibitors for medulloblastoma are complicated by the relatively small patient population (<30% of all medulloblastoma) and their overall relatively favorable prognosis.
154 Additionally, genomic alterations in several different genes (e.g., SUFU, PTCH1, GLI2) result in SHH pathway activation, and some of these genomic alterations (e.g., SUFU mutations and GLI2 amplification) are not responsive to Smoothened inhibitors.
19
Toxicity Concerns for the Use of Molecularly Targeted Agents in Children
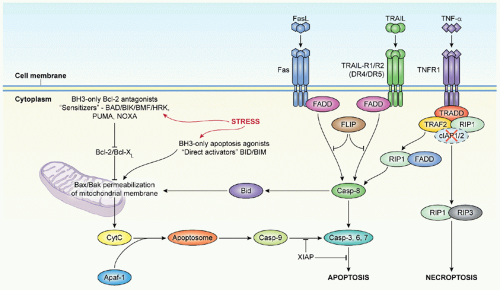
There are general issues about toxicities associated with molecularly targeted agents that apply to both adults and children. While presentations touting the promise of molecularly targeted agents often include reference to their lack of toxicity, this perception does not match reality for many “molecularly targeted” agents. Although these agents often lack the myelosuppression and gastrointestinal toxicities associated with cytotoxic agents that target rapidly cycling cells, many of the agents induce significant mechanism-based adverse events that affect their clinical development. For example, Notch pathway inhibitors cause diarrhea as a result of the goblet cell hyperplasia that occurs when Notch signaling is blocked.
127 Inhibitors of BCL-X
L cause a rapid-onset thrombocytopenia as a result of the dependence of platelets on BCL-X
L for survival in the circulation.
162,163 Cardiac toxicity has been associated with trastuzumab and with multitargeted receptor tyrosine kinases.
164 Some kinase inhibitors, including mTOR inhibitors, cause rashes and stomatitis that limit dosing. Thus, assumptions of lack of toxicity are misplaced for molecularly targeted agents, and as with conventional cytotoxic agents, the acute and long-term risks from their use will need to be carefully weighed against the evidence for their benefit.
The maxim that children are not simply young adults applies to the clinical development of molecularly targeted agents, just as it does during development of conventional cytotoxic agents. As with any therapeutic agent, children may tolerate molecularly targeted agents differently than adults because of age-related changes in physiology that result in pharmacokinetic differences between children and adults. In addition, molecularly targeted agents may specifically interfere with critical developmental pathways and block the progression from immature to mature adult tissues. For example, the central roles of the Wnt and Hedgehog signaling pathways in bone development will have to be considered as agents blocking these pathways enter clinical evaluation,
165,166 a point that was emphasized by the permanent defects in bone structure in young mice caused by transient inhibition of the Hedgehog pathway.
167The effect of VEGF inhibitors on the growth of immature animals illustrates the distinctive challenges for the pediatric development of molecularly targeted agents.
168 Vascularization of cartilage in long bones is observed during three periods in the life of vertebrates: during late embryonic development, during periods of rapid growth in immature animals as capillary structures invade at the growth plate regions, and during adulthood when angiogenesis is activated for bone remodeling in response to bone injury or other pathologic conditions.
169 Both antibodies that sequester vascular VEGF and small molecule inhibitors of VEGF receptors produce a characteristic effect on growth plates in immature mice and nonhuman primates.
168,170,171 In these animals, blood vessel invasion of growth plate cartilage is markedly suppressed, concomitant with impaired trabecular bone formation and marked expansion of the hypertrophic chondrocyte zone. Importantly, changes associated with relatively brief intervals of angiogenesis inhibition are reversible.
171Bone effects have been observed in a small number of children treated with VEGF-pathway targeted agents. An infant with cutaneovisceral angiomatosis with thrombocytopenia (CAT) syndrome
receiving bevacizumab developed asymptomatic metaphyseal bone lesions that reversed following cessation of bevacizumab.
172 Physeal widening was observed in the pediatric phase 1 trial of pazopanib.
173 An 11-year-old Tanner stage 2 patient showed a threefold expansion by cycle 10 of pazopanib as well as a decrease in height velocity. Three other patients on this trial (ages 11, 8, and 4 years) also showed radiographic growth plate widening after four cycles of pazopanib, but were removed as a result of progressive disease. That physeal widening has not been reported for other VEGF pathway inhibitors studied in children with cancer may reflect the small number of children studied and the limited duration of treatment and follow-up that is common for phase 1 clinical trials.
174,175,176 Three children with recurrent brain tumors treated with bevacizumab in combination with irinotecan developed osteonecrosis in the wrist or the knee, adding another bone toxicity to monitor for in children treated with VEGF pathway inhibitors.
177 The limited clinical experience with VEGF pathway inhibitors in children with cancer prevents the drawing of conclusions about the long-term impact of this class of agents on bone growth in children.
Growth retardation has been documented in children receiving long-term treatment with imatinib for CML.
178,179,180 The growth inhibitory effects of imatinib mesylate appear to be most pronounced in prepubertal children, compared with pubertal children,
178,179 and one study reported that growth velocity tended to recuperate in prepubertal children with growth impairment, as they reached pubertal age.
178 There is evidence for imatinib-induced growth failure in children that results from perturbations of the growth hormone (GH):IGF-1 axis, with reduced IGF-1 and IGFBP-3 levels and reduced response in growth hormone stimulation tests observed in children receiving imatinib.
180,181,182 Of note, second generation tyrosine kinase inhibitors such as dasatinib and bosutinib reduced IGFBP-3 levels in rats in a similar manner as imatinib, suggesting that the effects on the GH:IGF-1 axis may represent a class effect.
182 It is unknown whether GH replacement therapy for children with CML treated with imatinib is safe and effective at reversing growth retardation.
The acute and long-term effects of molecularly targeted agents described above highlight the need for careful monitoring of children receiving these agents. To date many children who have received molecularly targeted agents have had relapsed/refractory disease, and long-term survival was not expected. As more and more children with curative conditions receive these agents as part of frontline therapy (e.g., imatinib for CML, and Ph+ ALL and crizotinib for anaplastic large cell lymphoma), it will be particularly important to implement long-term follow-up programs so that late effects can be identified as promptly as possible and prevention and remedial actions taken.