Image 4.1
Adenocarcinoma, 10× magnification
Gender differences are present as well. Squamous cell carcinoma accounts for 44 % of lung cancers in men compared to 25 % in women, while adenocarcinomas comprise 28 % of lung cancers in men compared to 42 % in women. Frequencies of small- and large-cell carcinomas are roughly equal between men and women [7]. Part of the gender differences can be explained by the fact that a higher percentage of never-smokers are women [11, 12].
4.2.3 Treatment
The standard treatment for early NSCLC is surgery followed by adjuvant chemotherapy intended to prevent recurrences caused by micrometastases [2]. Advanced-stage or metastatic NSCLC is treated with a doublet chemotherapy regimen, for example, a doublet regimen, including cisplatin or carboplatin plus either docetaxel, paclitaxel, vinorelbine, gemcitabine, or irinotecan; other platinum combinations; or non-platinum combinations [1–4, 13–15]. Older patients or those with poor performance status may be treated with a single agent [14]. It has been shown that adjuvant chemotherapy in resectable NSCLC provides a 5–15 % absolute increase in overall survival [2, 16, 17].
4.2.4 Common Genetic Alterations in Lung Cancer
Various genetic alterations are often present in lung cancers, some of which may have specific prognostic or predictive value. The three most common genetic alterations found across all subtypes of lung cancer are loss of heterozygosity of 3p (LOH 3p), TP53 mutations, and PRb mutations [7, 18–20]. Up to 80 % of cases demonstrate LOH 3p, which is the location of tumor suppressor genes such as FHIT [7, 9]. The TP53 gene, which encodes the p53 tumor suppressor, is mutated in 50 % of NSCLC and 70 % of small cell carcinomas [7, 19]. Some alterations occur in specific histologic types. For example, squamous cell carcinomas frequently overexpress cytokeratins 5, 6, 13, 14, 16, 17, and 19 and infrequently harbor KRAS mutations [7]. In contrast, adenocarcinomas contain KRAS mutations in up to 30 % (especially in smokers) [10], contain TP53 mutations [10], and show overexpression of p27 [9, 21] and Cox-2 [7, 21].
Of the molecular alterations identified in lung carcinomas, some have been shown to have prognostic implications. Never-smoking status, high epidermal growth factor receptor (EGFR) protein expression, and increased EGFR gene copy number were all independent prognostic factors in the study by Hirsch et al. [22]. In squamous cell carcinomas, loss of PRb expression portends poor prognosis [7]. TP53 mutation has not been shown to have prognostic significance in squamous cell carcinomas but indicates poor prognosis for those with stage I and II adenocarcinomas [7, 19]. Poor prognosis may also accompany KRAS activating mutations and HER-2/neu overexpression [7]. Overexpression of p27 is correlated with well-differentiated tumors and indicates a good prognosis [7] (Fig. 4.1).
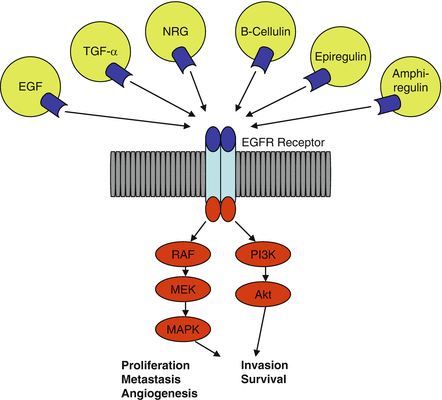
Fig. 4.1
EGFR Receptor involved in tumorigenesis of non-small cell lung cancer. EGFR epidermal growth factor receptor, EGF epidermal growth factor, TGF transforming growth factor, NRG neuregulin, RAF v-raf 1 murine leukemia viral oncogene homolog 1, Akt protein kinase B, PI3K, phosphatidylinositol-3-kinase, MEK, MAPK mitogen-activated protein kinase
4.3 Theory Behind the Use of Molecular Predictive Markers
Despite the fact that, in the population as a whole, chemotherapy provides a survival benefit for NSCLC, not every patient will respond; worse yet, many may only suffer from the adverse effects. This includes death in 1–2 % of patients receiving platinum-based regimens [17]. To avoid treating patients who are unlikely to respond, it would be ideal to have predictive markers to determine whether a patient would be likely to respond to a given regimen [1]. Such predictive markers may be positive, such as the presence of an activating EGFR gene mutation predicting response to tyrosine kinase [22], or negative, such as a KRAS mutation predicting resistance to tyrosine kinase inhibitors (TKIs). Predictive markers are not to be confused with prognostic markers or factors that estimate a patient’s likely outcome regardless of therapy [1].
Molecular alterations that serve as predictive markers are naturally going to fall into the category of a gene encoding drug metabolism, transmembrane transport, or target proteins [23]. Among the most studied predictive markers in lung cancer are EGFR, excision-repair cross complementing-1 protein (ERCC1), KRAS, and ribonucleotide reductase subunit 1 (RRM1). The following sections will discuss each of these markers, from the mechanisms by which they are thought to influence response to therapy and the clinical evidence supporting their value as molecular predictive markers.
4.4 Epidermal Growth Factor Receptor
4.4.1 Background
The most promising and widely reported molecular predictive factor in NSCLC is EGFR. The epidermal growth factor itself was first identified by Cohen in 1962 [24–26]. After epidermal growth factor binds to the EGFR, the receptor dimerizes, autophosphorylates, and activates several pathways, including MAP kinase, Jak2/STAT3, STAT5, and phosphoinositide 3-kinase (PI3K/Akt), which stimulate proliferation, metastasis, and migration and prevent apoptosis [5, 27–31]. Levels of phosphorylated STAT3 are most significantly correlated with phosphorylated EGFR levels, indicating that most of the signals from activated EGFR are transmitted through the STAT3 pathway [5]. It has also been shown that EGFR that is constitutively active as a result of either an exon 19 deletion or L858R substitution preferentially stimulates STAT and AKT pathways, preventing apoptosis but not promoting proliferation [28, 32]. This is consistent with the observation that there is no association between EGFR mutation and stage or metastasis, indicating absence of a link to tumor progression [32].
Because the epidermal growth factor normally favors growth and proliferation, mutations of its receptor that render it constitutively active independent of ligand-binding lead to a malignant phenotype [33, 34]. Increased EGFR gene copy number or overexpression has also been investigated for each of their roles in oncogenesis. It has been suggested that, based on their mechanisms of action, EGFR monoclonal antibodies (such as cetuximab, matuzumab, and panitumab, which bind the extracellular domain of EGFR and prevent ligand binding) should be beneficial to patients with tumors overexpressing surface EGFR, whereas TKIs should benefit patients having tumor proliferation and metastases driven by EGFR autophosphorylation [24, 35, 36]. In the latter case, blocking the catalytic site of the receptor (responsible for activating downstream molecules) could conceivably prevent growth and proliferation and even favor apoptosis in cancer cells. Two promising drugs that target the catalytic domain of the EGFR by competing with ATP are the reversible TKIs erlotinib and gefitinib [5, 36, 37]. Tumors likely to respond to TKIs are the ones that are dependent on aberrant signaling through the EGFR pathway for survival [36, 38].
The TKI erlotinib has been shown in a randomized, placebo-controlled, phase III trial to provide significant survival benefit compared to placebo in unselected, previously treated advanced-stage NSCLC patients [25]. In this trial and in others published before the discovery that lung tumors harboring mutations in the EGFR gene are sensitive to TKIs, it was observed that patients with certain demographic characteristics responded better to these agents. Studies had demonstrated that higher response rates to gefitinib were observed in women, Japanese patients, and in those with adenocarcinomas [11, 29, 39, 40]. The higher response rate in Japanese patients and the better outcome in women were not attributable to differences in pharmacokinetics in these populations [40]. It was also known that never-smokers had higher response rates, longer times to progression, and longer median overall survivals when treated with TKIs than smokers [11, 41]. These clinical characteristics (female sex, Asian origin, never-smoking status, and adenocarcinoma histology) have been independently associated with response to erlotinib [25, 42] and gefitinib (never-smoking status and female sex in Japanese patients) [43], and it has been suggested that these clinical characteristics may be of value as surrogate predictors of response to TKIs when molecular profiles are not available [44, 45]. Three articles published in 2004 revealed that EGFR mutations are the common factor linking the aforementioned patient demographics with consistent response to TKIs [7, 46]. Although erlotinib was shown to provide survival benefit as compared to placebo in a phase III trial [25], it was disappointing that gefitinib was shown only to be non-inferior to docetaxel in a randomized, placebo-controlled phase III trial, with no benefit over docetaxel even in subgroups of women, patients with adenocarcinomas, never-smokers, or Asian patients [47]. Compared to placebo, however, gefitinib did provide significant survival benefit for never-smokers and Asian patients in the randomized, placebo-controlled ISEL trial [48]. Without this subgroup analysis, survival was not significantly longer with gefitinib even compared to placebo, although response rate was significantly higher with gefitinib in the whole group and most pronounced in subgroups of patients with adenocarcinoma, never-smokers, women, and Asian patients [48].
EGFR activity may be increased in NSCLC by several different mechanisms, which may be assessed separately in the lab. The gene, EGFR (located on chromosome 7p12), may be mutated and/or amplified and the EGFR protein may be overexpressed in the presence or absence of either of these abnormalities. Background information and clinical studies regarding the predictive value of each of these aberrations are reviewed in the following sections.
4.4.2 EGFR Mutations
4.4.2.1 Types of Mutations
Many different types of mutations have been identified in the EGFR gene, approximately 90 % of which are either small deletions from exon 19 (codons 746–750) or substitution of arginine for leucine at position 858 in exon 21 (L858R). Another 3 % are substitutions of a variety of amino acids in place of glycine at codon 719 (G719X) and 3 % are in-frame insertions in exon 20 [7, 29, 37, 46, 49–51]. The overall incidence of EGFR mutations in NSCLC ranges from 12.1 % to 49 %, depending on the patient population [37, 46, 49–51]. Tyrosine kinase domain mutations of EGFR are rare in non-lung tumors [28].
The exon 19 deletions and L858R substitution are activating mutations that result in increased phosphorylation of EGFR independent of ligand stimulation [11, 28, 29, 52]. These mutant EGFRs have greater affinity for TKIs and are susceptible to inhibition by TKIs in vitro [34, 52]. Politi et al. and Ji et al. each showed that expression of either the exon 19 deletion or L858R substitution mutation in EGFR causes adenocarcinoma to develop in mice and that the tumors regressed when either expression of the mutant gene was blocked or erlotinib was administered [33–35, 53]. Greulich et al. obtained similar results in cell cultures and confirmed that these mutations result in constitutive ligand-independent receptor activation, which is further enhanced by the addition of ligand stimulation [54]. In summary, EGFR genes specifically with activating mutations produce EGFR that demonstrates ligand-independent stimulation of downstream growth and survival pathways that produce the malignant phenotype [5, 11, 33–35, 53, 55].
4.4.2.2 Clinical Characteristics of Patients with EGFR Mutations
Since the discovery that NSCLCs with EGFR mutations may be more responsive to TKIs, the characteristics of patients and tumors harboring these mutations have been validated again and again. A compilation of data from 2,880 patients generated in the review by Mitsudomi et al. in 2007 shows that EGFR mutations are found in 32 % of Asians compared to 7 % of non-Asians, 38 % of females compared to 10 % of males, 47 % of never-smokers compared to 7 % of smokers, and 30 % of adenocarcinomas compared to 2 % of non-adenocarcinomas [29]. Many other studies have also found EGFR mutations to be significantly associated with females, Asian origin, never-smoking status, and adenocarcinoma histology [22, 36–38, 41, 42, 45, 46, 51, 56–58], which overlap with characteristics of patients who respond to TKIs [25]. From a different angle, smoking has been shown not to be a risk factor for EGFR-mutant NSCLC, whereas female sex is a significant risk factor [59]. A significant association between presence of EGFR mutation and younger age was even demonstrated by Eberhard et al. in the phase III TRIBUTE study [49]. Compared to white patients, African Americans are significantly less likely to have activating mutations of EGFR [60]. Among Korean patients with adenocarcinomas, the presence of EGFR mutations has not been shown to be associated with gender, smoking history, histological grade, age, bronchioloalveolar component, or stage [61]. Despite the statistical associations, it is important to remember that there will be exceptions to the rules. Park et al. in 2009 found that 3 of 20 male patients with squamous cell carcinoma of the lung had EGFR mutations, all of which responded to gefitinib, and, in addition, one of the EGFR-wild-type patients responded as well [38].
4.4.3 Histology of EGFR-Mutant Tumors
With respect to histology, it has been shown that, among adenocarcinomas that contain EGFR mutations, the majority are of terminal respiratory unit type [28, 62]. This subset of lung adenocarcinomas is characterized by cytologic resemblance to type II pneumocytes, Clara cells, or nonciliated bronchiolar cells [62]. Positive immunohistochemical staining for both thyroid transcription factor-1 and surfactant precursor protein B is a highly sensitive and specific method of identifying TRU-type lung adenocarcinomas [62]. EGFR mutations have also been identified in areas of atypical adenomatous hyperplasia, a preneoplastic lesion [62].
4.4.4 Relationship of Smoking Status to EGFR Mutations
The first molecular alteration that is actually found more often in non-smokers with NSCLC than in smokers is mutation of EGFR [28, 35]. Although some studies have shown a significant relationship between EGFR mutation presence and non-smoking status [6, 45, 56, 63, 64], and others have shown a trend toward lower EGFR mutation incidence with higher cumulative smoke exposure [29], not all studies have found a significant relationship between presence of EGFR mutation and smoking status [49, 61]. Even though a given never-smoker may be more likely to have an EGFR mutation than a smoker, EGFR mutations are still occasionally identified in smokers. Mitsudomi et al. in 2007 found EGFR mutations in 20 % of NSCLC from smokers, explaining that the reason EGFR mutations are found in a smaller percentage of tumors from smokers is that they are diluted by tumors arising from different smoking-induced mechanisms not usually present in never-smokers [30, 31, 35, 59]. To further investigate the relationship between smoke exposure and EGFR mutation status, Lee et al. examined the incidence of EGFR mutations in never-smokers with and without environmental tobacco smoke exposure, finding a significantly lower incidence of EGFR mutations in those with environmental tobacco smoke exposure than in those without. They also found a trend toward lower response to TKIs in patients with environmental tobacco smoke exposure compared to those without [64].
4.4.5 EGFR Mutation as a Molecular Predictor of Response to TKIs
The heart of determining the usefulness of EGFR mutation status as a predictive factor for response to TKIs lies in the clinical trials and retrospective studies investigating this relationship. In 2004, Lynch et al. was one of the first groups to demonstrate that EGFR mutations are associated with clinical response to gefitinib and to show in vitro that EGFR mutants were more sensitive to gefitinib. No mutations were present in patients who did not respond to gefitinib. In vitro, both the exon 19 deletion mutants and L858R mutants were more sensitive to gefitinib than tumors with wild-type EGFR. All eight mutants they identified were heterozygotes, indicating that these mutations act in a dominant manner (as do other oncogenes) [46]. Paez et al. promptly confirmed these results [41] and Pao et al. soon added that EGFR mutations were also identified in tumors sensitive to erlotinib [45]. Since then, many studies have confirmed association between EGFR mutation status and response to TKIs, and a review of the literature by Mitsudomi et al. in 2007 compiling data from 1,335 patients showed that EGFR TKI response was observed in 70 % of EGFR mutant tumors compared to only 10 % of patients with EGFR wild-type tumors [15, 29, 43, 58, 63, 65, 66]. In the phase II IDEAL trial, EGFR mutant tumors responded better to gefitinib than those with wild-type EGFR [58]. Kalikaki et al. demonstrated that patients with classic activating EGFR mutations treated with gefitinib had significantly longer median overall survival than those with wild-type EGFR. This relationship was not present in patients with other types of EGFR mutations [55]. Significantly higher objective tumor response to TKIs in EGFR mutants compared to wild-type has been demonstrated by several studies [37, 50, 65, 67], although others have found no relationship [42].
Differential response to treatment between the different types of EGFR mutations have been observed by some authors. Mitsudomi et al. reported that deletional mutations had higher response rates to gefitinib than other mutations, especially L858R [29]. Along those lines, Hirsch et al. found that patients who only had exon 19 deletions had significantly higher objective response than patients with exon 21 mutations [22]. On the contrary, Pao et al. found in vitro that wild-type EGFR and exon 19 deletion mutants responded similarly to gefitinib and erlotinib, whereas L858R mutants were about 10 times as sensitive [45]. As of yet, there has been no report of response to TKIs in a patient with an exon 20 insertion [29].
Not all patients with activating mutations of EGFR respond to TKIs [68]. Conversely, some patients without EGFR mutations still respond to TKIs [7, 67, 68]. In 2009, Yoshioka et al. published a phase II prospective study with EGFR mutation status determination followed by erlotinib treatment and determined that EGFR wild-type tumors have “modest” response to erlotinib without irreversible toxicity [69]. Some of the mechanisms by which patients harboring EGFR mutations become resistant to TKI therapy are discussed later in this chapter.
4.4.6 EGFR Mutation as Molecular Predictor of Response to Chemotherapy
TKIs are not FDA approved for use as first-line therapy for NSCLC in the United States [36]. Because first-line therapy is limited to conventional chemotherapy (a doublet regimen: for example, cisplatin or carboplatin plus either docetaxel, paclitaxel, vinorelbine, gemcitabine, or irinotecan; other platinum combinations; or non-platinum combinations) [2–4, 13, 14] (as mentioned previously), naturally the impact of EGFR mutation status on response to initial chemotherapy has been investigated. Results of such studies have been conflicting, with many studies reporting no association, while Hotta et al. showed that EGFR mutations were associated with longer overall and progression-free survival but not better response to front-line chemotherapy (consisting of various regimens, including both platinum- and non-platinum based) and other studies showed that only patients who carried deletions in exon 19 of EGFR had response to initial chemotherapy [15, 55]. Kalikaki et al. found a higher objective response rate to initial chemotherapy (particularly platinum-based regimens) in patients with classic activating EGFR mutations compared to wild-type EGFR [55]. First-line cytotoxic chemotherapy response was not correlated with EGFR mutation status in the study by Yoshida et al. (the exact chemotherapy regimens were not specified in this study) [69].
4.4.7 TKIs Versus Chemotherapy in EGFR Mutant Tumors
Although many studies have shown a benefit of erlotinib over placebo in EGFR-mutation positive patients, differential responses between TKIs and conventional chemotherapy have been more difficult to demonstrate. In the phase III TRIBUTE study, erlotinib did not provide any survival, objective response rate, time to progression, or duration of response advantage over carboplatin plus paclitaxel alone in unselected patients [44, 49]. Similarly, gefitinib did not provide any survival advantage compared to docetaxel in unselected patients or subgroups with clinical characteristics associated with EGFR mutations or EGFR mutations themselves in the phase III INTEREST trial [47]. However, patients with EGFR-mutant tumors had significantly improved objective response rate compared to wild-type tumors when treated with erlotinib plus carboplatin plus paclitaxel in the TRIBUTE study [49]. There was even a nonsignificant trend toward better response to erlotinib plus carboplatin plus paclitaxel compared to carboplatin plus paclitaxel alone in EGFR-mutant patients [49]. Regardless of treatment arm in this study, patients with EGFR-mutant tumors had significantly better response rate and longer median time to progression than those with wild-type tumors [49]. The randomized, placebo-controlled INTACT trials did not show added benefit from gefitinib in addition to chemotherapy alone in unselected patients [58]. In at least one study, higher objective response rate was observed in EGFR mutation-positive patients treated with gefitinib compared to docetaxel, although mutation status did not predict better survival [70].
4.4.8 Impact of EGFR Mutation on Survival
Impact of EGFR mutation status on treatment response is of great importance, but may have little meaning to patients unless there is a concomitant impact on survival. Longer median survival (but not progression-free survival) after first-line chemotherapy was observed in patients with EGFR mutations [71]. Overall and progression-free survival following first-line chemotherapy were also significantly longer in patients with EGFR-mutant tumors than in those with wild-type EGFR in the study by Hotta et al. [15]. With respect to TKI (gefitinib) treatment, use of this agent as first- or second-line therapy resulted in significantly longer progression-free survival in EGFR-mutant patients than in those treated with chemotherapy [71]. In a series of 397 patients by Kosaka et al. in 2009, patients with EGFR mutations survived significantly longer than those with wild-type mutations, with no survival difference between exon 19 deletion and L858R substitution [51].
4.4.9 Molecular Prognostic Value of EGFR Mutations
Because subset analyses of the TRIBUTE and INTACT trials have shown that better response was observed in patients with EGFR mutations patients even on chemotherapy alone (not just with chemotherapy plus TKI), it has been argued that EGFR mutations are prognostic factors instead of predictive [35, 49, 58]. Other studies have confirmed that patients with EGFR mutations (or clinical characteristics classically associated with EGFR mutations) have longer survival on chemotherapy even without TKIs [47, 49, 55, 58, 71]. Marks et al. found a trend toward longer overall survival in patients with EGFR mutations and stage I disease compared to patients with EGFR wild-type or KRAS-mutant tumors [72]. On the other hand, both Kosaka et al. and Shigematsu et al. showed that EGFR mutation was not a significant prognostic factor in patients treated with surgery alone [35, 51, 57]. In 2009, Kosaka et al. again showed in their series of 397 patients that neither the exon 19 deletion nor the L858R substitution mutation was an independent prognostic factor [51]. It has been suggested that, because of the characteristics of patients who tend to have EGFR mutations, they have better prognosis regardless of treatment given [7, 47].
4.4.10 Sensitivity of EGFR Mutation Detection Methods
As important as detection of EGFR mutations may be for patients with NSCLC, currently implemented detection methods may be missing a large portion of positive cases. Hirsch et al. compared two methods to detect EGFR mutations: direct sequencing and amplification refractory mutation system analysis (ARMS). ARMS uses allele-specific PCR to detect the exon 21 L858R point mutation and the most common exon 19 deletion (del G2235-A2249). They concluded that ARMS is more sensitive than sequencing (for detecting the most common mutations) because 10 of the 17 mutations detected by ARMS were missed by sequencing. However, since sequencing is not restricted to detecting specific known mutations, it identified overall 10 different mutations that were missed by ARMS; therefore, it is useful in detecting novel or uncommon mutations [37].
4.5 EGFR Gene Copy Number
4.5.1 Background and Clinical Associations with EGFR Amplification
EGFR gene copy number has also been investigated for its possible predictive and prognostic values in NSCLC. In general, a gene’s copy number may be increased as a result of true amplification or it may accompany polysomy of its entire chromosome. Gene copy number in either case is determined using fluorescence in-situ hybridization (FISH) [73, 74]. The frequency of FISH positivity for EGFR ranges from 30 % to 50 % in NSCLCs and is more frequent in older patients and women in some studies [22, 37, 50, 58, 60, 63, 70]. In the IDEAL and INTACT trials, the demographic characteristics that are similar between patients with EGFR mutations and those that respond to TKIs (female, never-smokers, adenocarcinoma, Asian origin) were not associated with EGFR amplification [58]. In one study, EGFR FISH positivity was observed significantly more often in African Americans than whites, which was entirely attributable to the cases with high polysomy [60]. Never-smokers have been shown to more often have high EGFR gene copy numbers than smokers [22, 63, 70].
4.5.2 Relationship Between EGFR Mutation and Amplification
Scholl et al. reported on a significant relationship between EGFR gene amplification and exon 19 deletions [75]. In a study of 99 pulmonary adenocarcinomas from chemotherapy-naïve, never-smoking east Asian women, significantly more exon 19 deletions were present in tumors with EGFR amplification than in those with EGFR disomy or low or high polysomy [75]. Further, only the mutated, not the wild-type allele, was amplified [75]. They concluded that EGFR amplification only occurs in tumors with exon 19 deletions of EGFR and not other types of mutations or wild-type EGFR [75]. Other studies have found other mutations associated with EGFR amplification; however, in patients treated with prior chemotherapy [75], gene amplification is more often associated with EGFR mutations than is polysomy [73]. They further reported that, in tumors with exon 19 deletions, constitutive activation of EGFR occurs only in the presence of amplification [75]. In cells with the L858R substitution, constitutive activation was observed regardless of gene amplification [75]. Other studies have confirmed association between EGFR mutation of unspecified type and EGFR FISH positivity [63, 66, 67]. Increased EGFR expression by immunohistochemistry (IHC) was also significantly associated with FISH positivity but not mutation status [63, 76].
4.5.3 EGFR FISH: Criteria and Special Considerations
Varella-Garcia et al. have proposed the following criteria to define FISH positivity for EGFR: at least 40 % of the cells must have at least 4 copies of the EGFR signal, or the EGFR to centromere enumeration probe for chromosome 7 (CEP7) ratio must be at least two (averaged over all nuclei), or there must be gene clusters (at least 4 spots) in at least 10 % of tumors cells, or there must be at least 15 copies of the EGFR gene in at least 10 % of the tumor cells [74]. The result is equivocal if none of the above criteria is met but at least 25 % of the cells have at least four signals; in this case, if a second reviewer calls it positive, it is reported as positive; otherwise the test is reported as negative [74].
Several reports state that TKIs may benefit patients with either EGFR amplification or 7p polysomy. However, gene amplification is more often associated with EGFR mutations than is polysomy [73]. Since the two mechanisms of increased EGFR gene copy number may have different predictive values, a method has been proposed to more reliably distinguish true gene amplification from chromosome 7 polysomy. The EGFR gene itself lies on chromosome 7p12, which is close enough to the centromere of chromosome 7 that gene amplification of this region may also amplify parts of the centromere, giving the false impression that the whole chromosome has been duplicated (polysomy 7). The 7q31 region is sufficiently far from both the centromere and the EGFR locus that identification of this region may help distinguish signals from EGFR amplification and signals from the centromere [73].
4.5.4 Molecular Predictive Value of EGFR Gene Copy Number Versus Mutation
Most of the studies assessing the predictive value of EGFR amplification compare this to the predictive value of EGFR mutation. Capuzzo et al. were the first to report that survival following gefitinib was best predicted by increased EGFR gene copy number compared to EGFR mutation status or EGFR expression by IHC [29, 44, 63]. At least three other groups found EGFR gene copy number to be a better predictor of response to TKIs than EGFR mutation status [29, 35, 37, 42, 44, 71, 77]. In one study, EGFR FISH positivity was even associated with significant survival benefit from erlotinib, which was not observed for tumors with wild-type EGFR, mutant EGFR, or those that were EGFR FISH-negative [50]. A significantly better response to gefitinib compared to docetaxel was seen in patients with high EGFR gene copy number in another study, although gene copy number was not associated with better survival in this study [70].
In the Hirsch study, patients whose tumors were EGFR negative by both FISH and IHC had poor response to gefinitib regardless of EGFR mutation status. Inversely, patients positive for EGFR by both FISH and IHC had excellent outcome independent of EGFR or KRAS mutations. These findings suggest that EGFR gene copy number is a better predictor of response to gefitinib than EGFR mutation status [22, 35, 37, 71]. Hirsch et al. determined that the combination of EGFR gene copy number and protein expression by IHC is the most predictive of response to gefitinib and concluded that “EGFR gene copy number is a predictor of survival benefit with gefitinib compared with placebo” [22, 37]. Takano et al. found after multivariate analysis that response to gefitinib in Japanese patients could be predicted by either high EGFR gene copy number or EGFR mutation alone but pointed out that the significance of increased copy number is clouded by the observation that mutation presence predicts response whether increased copy number is present or not [67].
On the other hand, at least three groups found EGFR mutation status to be a better predictor of response to TKIs than EGFR gene copy number [43, 66, 68, 71]. Variations in the demographics of the study populations may be partly responsible for the conflicting results. In fact, Gerber has pointed out that EGFR gene copy number seems to have more predictive value in Caucasian populations (who have low EGFR mutation frequency) [42, 63], whereas EGFR mutation status holds more predictive value in East Asian populations (who have higher EGFR mutation frequency) [36, 42, 58, 63, 67].
Not all studies have found evidence to support the predictive role of EGFR FISH positivity. Both the INTEREST and INVITE phase III studies failed to show association of EGFR FISH positivity with prediction of benefit from gefitinib compared to chemotherapy [47, 60, 78]. Although EGFR FISH positivity was associated with TKI response in univariate analysis in the studies by Han et al. in 2006 and Ichihara et al. in 2007, in multivariate analysis, the only factor that independently predicted both response and survival from gefitinib was gefitinib-sensitive EGFR mutation [35, 43, 66, 71]. Further, while EGFR FISH positivity may predict response to TKIs, it has no predictive value for response to chemotherapy [76].
4.5.5 Molecular Prognostic Value of EGFR FISH Positivity
In contrast to its association with favorable response to TKIs, EGFR FISH positivity may be a negative prognostic factor [7, 63, 76]. Zhu et al. in 2008 found that EGFR FISH positivity was a poor prognostic marker by multivariate analysis [50]. Sholl et al. also found significantly worse outcome (shorter disease-free survival and higher stage at presentation) in patients with EGFR amplification [75]. In the Hirsch study, there was a nonsignificant trend toward worse outcome in patients with high EGFR gene copy number compared to low gene copy number in the placebo group [22]. To reiterate, this is in contrast to the better outcome predicted by EGFR FISH positivity in patients treated with gefitinib [37]. Tsao et al. and Dziadziuszko et al. did not find EGFR gene copy number to have prognostic value [42, 76]. However, at least one study has suggested a contradictory prognostic implication of EGFR FISH positivity [77].
4.6 EGFR Protein Expression Levels
4.6.1 Background
The last mechanism of increased EGFR activity to be discussed is protein overexpression, determined using IHC. Up to 90 % of NSCLCs strongly express EGFR protein by IHC [37, 70, 79], more frequently in squamous cell carcinomas than in adenocarcinomas [39, 40, 76], which is notably the opposite histologic association found with EGFR mutations. Although the overexpression of wild-type EGFR does not result in the malignant phenotype [31, 53], and many studies have not demonstrated a relationship between EGFR protein expression and response to TKIs [27, 39, 47, 79, 80], other studies have shown better response to TKIs in tumors expressing EGFR by IHC [7, 22, 37, 42, 44, 50, 63].
Ethnicity-associated EGFR genotypes may underlie some of the predictive value of EGFR overexpression. EGFR gene transcriptional activity and EGFR expression are related to the number of CA repeats contained in intron 1, with a higher number of repeats correlating with lower EGFR transcription, lower levels of EGFR mRNA, and lower EGFR protein expression [7, 81, 82]. The 20-repeat allele has only approximately 20 % the EGFR transcription activity of the 16-repeat allele [7, 81, 83]. Assuming for a moment that EGFR protein expression may affect response to TKIs, differences in the number of CA repeats may account for some of the racial differences in response to TKIs in EGFR-positive tumors; for example, higher response rates in Japanese patients treated with TKIs may be because in Asia the 20-repeat allele is the most common, whereas Caucasians and African Americans more commonly have the 16-repeat allele [7, 61, 83].
4.6.2 Molecular Predictive and Prognostic Value of EGFR Overexpression
Based on evidence that activating mutations or amplification of EGFR predicts response to TKIs, one may suspect that EGFR overexpression may have similar predictive value. Indeed, Tsao et al. found expression of EGFR by IHC to be associated with response to erlotinib but not with survival [42]. Parra et al. also found that, within a subset of patients with adenocarcinoma, response to gefitinib was significantly associated with high EGFR expression by IHC [39]. Whereas some authors have suggested that EGFR overexpression by IHC may predict response to TKIs, the evidence that overexpression may be related to response to conventional chemotherapy is limited [76, 84]. Comparing response to a TKI and conventional chemotherapy, there was no differential survival between gefitinib treatment and docetaxel treatment with respect to EGFR expression status in the phase III INTEREST trial [47]. Evidence for any predictive value of EGFR overexpression is sparse; in fact, it has been reported that EGFR overexpression actually correlates more with the occurrence of skin toxicity resulting from TKIs than to response of the tumor [7]. Although at least one study demonstrated significant poor prognostic value of high EGFR expression by IHC [39], neither the predictive nor the prognostic value of EGFR protein expression has been established [39, 42, 63, 76, 79, 80, 85].
Results of studies comparing EGFR expression to therapy response are dependent on the method of protein detection (i.e., the specific antibody used), and such differences between studies may account for some of the conflicting results [39, 79]. Comparing results from four different antibody kits, Mathieu et al. found three kits to yield 80 % positivity in a series of cases, while the fourth kit yielded only 68 % positivity [79]. Additionally, biopsies were more often positive than surgical or cytology specimens using one particular kit, while metastases were more often negative than primary tumors using another kit [79].
4.7 EGFR Summary
Over time, differing opinions have existed on whether there is enough data to confirm the predictive value of EGFR mutations. In 2005, the authors of the TRIBUTE study believed that the use of EGFR mutation status as a marker predicting benefit to erlotinib plus chemotherapy was not clear because, although EGFR mutants had significantly better objective response rates than wild-type EGFR when treated with erlotinib plus carboplatin and paclitaxel, adding erlotinib to carboplatin plus paclitaxel did not improve response in EGFR mutants compared to carboplatin and paclitaxel alone [49]. Later publications have stated a more definitive predictive value, which likely arose from more and more data confirming the superiority of EGFR mutational analysis over IHC and FISH. In 2006, Han et al. found that, in multivariate analysis, the only factor that independently predicted both response and survival from gefitinib was gefinitib-sensitive EGFR mutations [35, 66]. Kalikaki et al. in 2009 went as far as to show that classical activating mutations of EGFR are an independent factor predicting response to first-line chemotherapy [55]. Thus, although amplification and overexpression of EGFR may have some predictive value, their analysis has not been employed clinically as often as mutation analysis.
Even though the presence of an activating mutation of EGFR may indicate that a patient is more likely to respond to TKIs, some patients without mutations do respond. With this in mind, a patient with a good performance status who does not respond to first-line chemotherapy may wish to pursue TKI therapy regardless of EGFR mutation status. EGFR testing can also help with estimating prognosis in some patients.
Testing is not warranted in all patients, especially when the test result (e.g. EGFR mutation status) is not going to change clinical management (e.g., administration of a TKI). Other patients who may not require testing for EGFR mutations include patients who come for second opinions and are already taking TKIs and showing response, patients with such poor performance status and for whom systemic therapy would not even be considered, and patients in whom initial biopsy material is insufficient for testing and risk of re-biopsy outweighs benefit. In addition, difficulties arise for patients who lack insurance or have insurance issues and thus may have difficulties paying for testing and/or treatment (Dr. Jhanelle Gray, telephone interview). For these types of issues, individuals should seek help from assistance programs.
In summary, patients with activating EGFR mutations are more likely to respond to TKI therapy and probably have improved prognosis independent of therapy.
4.8 Tyrosine Kinase Inhibitors
4.8.1 Adverse Effects of Tyrosine Kinase Inhibitors as Predictors of Response
As might be expected from its name, epidermal growth factor is critical for mediating differentiation and proliferation of keratinocytes, explaining why a rash often develops with its inhibition [31, 86, 87]. In some studies, the development of acneiform rash has been quite convincingly correlated with better tumor responses and improved overall and progression-free survival with TKI treatment, likely because skin toxicity is related to systemic drug concentration and activity [5, 80, 86, 88, 89]. There has also been a significant correlation between grade of rash and degree of tumor response to erlotinib [89]. Accordingly, some authors consider skin toxicity to be a predictive marker of response to TKIs [88].
4.8.2 Molecular Alterations Conferring Resistance to TKIs
Although many tumors with EGFR mutations initially respond to TKIs, disease progression invariably occurs (usually within 6–12 months), commonly by selective survival of tumor cells with acquired mutations conferring resistance to these agents [29, 35, 90, 91]. Even small clones of tumor cells containing a mutation that confers resistance to TKIs may have a significant clinical impact [91, 92]. The following section discusses the mechanisms behind several molecular alterations that unfortunately predict resistance to TKI therapy.
4.8.2.1 T790M
The most common secondary EGFR mutation leading to TKI resistance is substitution of methionine in place of threonine at codon 790 (T790M), independently identified by Pao et al. and Kobayashi et al. in 2005 [35, 90, 93–95]. After identifying the presence of the T790M point mutation in a patient who was initially sensitive to gefitinib but later became resistant, Kobayashi et al. demonstrated in vitro that introducing the T790M mutation into previously gefitinib-sensitive NSCLC cells harboring either wild-type, exon 19 deletion, or L858R substitution EGFR genotypes induced resistance to gefitinib [94]. Pao et al. also demonstrated in vitro that NSCLC cells harboring the T790M mutation with otherwise wild-type or mutated EGFR (either exon 19 deletion or L858R substitution) did not respond to either erlotinib or gefitinib [93].
Structural analysis performed by Kobayashi et al. showed that the T790M point mutation of EGFR changes the tyrosine in the catalytic domain to methionine, which is bulkier and prevents erlotinib (and presumably gefitinib) from binding [7, 35, 94, 96]. Not only does the T790M mutation alter TKI binding, but it also enhances kinase activity of EGFR (when coupled with the L858R mutation), offering cells with this mutation a survival advantage [11, 29, 52, 97]. Another missense mutation, D761Y, has been reported to confer a milder degree of TKI resistance, which may still be clinically relevant [98]. Greater resistance to gefitinib is produced when the T790M mutation occurs on the same DNA strand as the EGFR activating mutation (L858R or exon 19 deletion) [99], which is the most common scenario [100]. This can be explained by understanding that only the mutant allele will code for the mutant, constitutively active EGFR; likewise, binding of TKIs to these mutant, constitutively active EGFRs will only be altered if the resistance mutation has occurred on the mutant allele.
The T790M point mutation has been identified in approximately 50 % of patients who had activating EGFR mutations and later acquired resistance to EGFR TKIs [35, 44, 91, 95, 96, 98]. Engelman et al. demonstrated in vitro that malignant cells with activating mutations of EGFR that are initially sensitive to gefitinib become resistant after prolonged exposure to gefitinib and may acquire the T790M mutation in EGFR [44, 99]. Even so, the question arises as to whether TKI treatment itself induces the resistance mutation or if it merely results in selection of resistant clones [96]. Considering that the T790M mutation is found in 0.5–3.6 % (depending on the sensitivity of the assay; highest with mutant-enriched PCR) of patients who have never been treated with TKIs [29, 91], it is likely that TKIs merely result in selection of resistant clones rather than induce the T790M mutation themselves [29, 91].
4.8.2.2 Exon 20 Insertion Mutation
The exon 20 insertion mutation of EGFR (D770insNPG) has been demonstrated by Greulich et al. to confer erlotinib and gefitinib resistance in vitro. Response was obtained, however, from the irreversible EGFR inhibitor CL-387,785 [54]. Two patients harboring this mutation (who incidentally were both never-smoking women with adenocarcinomas) both progressed despite gefitinib treatment [66]. Intermediate sensitivity to erlotinib or gefitinib was observed in G719S of exon 18. The authors suggested that sensitivity may be related to the specific mutation present [35, 44, 54].
4.8.2.3 Low PTEN Expression
Low phosphatase and tensin homolog (PTEN) expression is a major reason why tumors become resistant to TKIs [101, 102]. PTEN normally functions to down-regulate the PI3K pathway (downstream of EGFR; mediates growth, proliferation, and survival). Loss of PTEN leads to persistent activation of the PI3K pathway independent of EGFR signaling, making this proliferation signal unresponsive to EGFR blockade [101, 102]. Bianco et al. demonstrated that, in gefitinib-resistant cells lacking PTEN, reconstitution of PTEN reversed EGFR-independent PI3K pathway activity and restored gefitinib sensitivity [102]. Zhuang et al. demonstrated that irradiating TKI-resistant tumors with low PTEN expression may help reverse the resistance by increasing PTEN expression levels [101]. Although low PTEN expression may induce resistance to TKIs, there may not be a relationship between PTEN expression and survival [27].
4.8.2.4 MET Amplification
Some authors have reported acquired resistance to EGFR TKIs in tumors with MET amplification [29, 99, 103]. It has also been suggested that MET activation (which may be induced by hepatocyte growth factor binding, overexpression, or structural alterations) may be used in place of MET amplification to determine sensitivity to TKIs [103]. The results of an in vitro study by Rho et al. suggested that sensitivity to TKIs was not associated with MET activation in the absence of MET amplification [103].
4.8.3 Methods to Overcome Resistance Mutations
Methods to overcome T790M-induced EGFR TKI resistance are of clinical interest. Murine and in vitro studies reveal some agents that may be beneficial. Although T790M makes cells resistant to gefitinib and erlotinib (which are reversible TKIs), the effect of irreversible TKIs (which covalently bind the EGFR kinase domain) is not changed by T790M. Irreversible TKIs include EKB-569, HKI-272, CI-1033, CL-387,785, and BIBW2992 [29, 94, 96, 98, 99]. The irreversible EGFR inhibitor CL-387,785 inhibits EGFR and downstream molecules more potently than gefitinib or erlotinib in cells harboring the T790M mutation, although not nearly to the degree that would be observed in cells without the T790M mutation [94, 99, 103]. Regales et al. demonstrated in mice that tumors harboring the T790M TKI resistance mutation had the most shrinkage (and complete response in most tumors) when treated with a combination of two drugs: BIBW-2992 (which is an irreversible EGFR TKI) and cetuximab (which is an EGFR-specific antibody). This combination resulted in lower total and phosphorylated EGFR. Neither agent alone induced complete response in any of the tumors in the study [90]. Another study has shown that, in mouse xenograft models of NSCLC with EGFR T790M mutations, CUDC-305 (a heat-shock protein 90 inhibitor) inhibits tumor growth [104].
Blocking signaling molecules downstream of EGFR has the potential to overcome resistance induced by a variety of mutations that alter binding of drugs to EGFR [53, 105]. Src is one such downstream signaling molecule that is vital to maintaining the malignant phenotype of EGFR mutant cells [53]. In vitro, Src inhibitors have been shown to prevent oncogenesis caused by EGFR mutations, suggesting that these may be of clinical benefit in TKI-resistant EGFR-mutated NSCLCs [53]. Faber et al. demonstrated in vitro that blocking two major pathways downstream from EGFR resulted in tumor shrinkage even in cell lines with various TKI resistance mutations including T790M [105].
4.9 Excision-Repair Cross-Complementation Group 1 (ERCC1)
4.9.1 Background
Whereas most of the discussion on molecular predictive factors in NSCLC has focused on response to TKIs, the ERCC1 enzyme may predict resistance to platinum-based therapy [7, 106–109]. Overexpression of ERCC1 occurs in 31–68 % of NSCLCs and is significantly associated with older age, squamous cell rather than adenocarcinoma histology, and pleural invasion [2, 13, 106, 110–112]. ERCC1 repairs platinum-DNA adducts (which cause inter- and intra-strand cross links) caused by platinum-based agents in cancer cells [7, 13, 106, 112–114]. This enzyme first recognizes the DNA damage and then removes the damaged nucleotides [115]. If the defects caused by the cytotoxic agent are not repaired, the cell will undergo apoptosis, which is, of course, the goal of administration of anti-cancer therapy. Through its DNA-repair action, ERCC1 reverses damage caused by certain chemotherapeutics on cancer cells, thus providing resistance to their anti-cancer effects [13, 113, 114, 116]. It is the expression level of ERCC1 that has been studied for its predictive value, since it has been established that ERCC1 mRNA levels correlate with DNA repair capacity [117].
4.9.2 Evidence for Molecular Predictive Value of ERCC1
Based on its normal biological function, ERCC1 has been studied for its relationship to chemotherapy resistance. In a phase III trial, patients were prospectively randomized to either a control arm, where all patients were treated with docetaxel plus cisplatin regardless of ERCC1 mRNA level, or a genotypic arm, where patients with low ERCC1 mRNA received docetaxel plus cisplatin. Those with high ERCC1 mRNA received docetaxel plus gemcitabine. The objective response rate was significantly higher in patients assigned to the genotypic arm than in those in the control arm group [65, 118]. It is interesting to note that patients in the genotypic arm responded better, even though gemcitabine was used for patients with high ERCC1 expression, which is correlated with high RRM1 expression. High RRM1 expression has been shown to predict resistance to gemcitabine treatment (to be discussed further in the following section) [118]. The results of this study clearly demonstrate the clinical utility of ERCC1 mRNA levels for determination of appropriate chemotherapy regimen for patients with advanced NSCLC [118].
Related posts:
 Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
 Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Cutaneous Malignancy
Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Cutaneous Malignancy
 Molecular Diagnosis of Bladder and Kidney Cancer
Molecular Diagnosis of Bladder and Kidney Cancer
 Molecular Pathology and Diagnostics of Childhood Tumors
Molecular Pathology and Diagnostics of Childhood Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


