Chromosomal defect
Childhood tumor
1p del, 1p32-p36 del, double minutes
Neuroblastoma
11p13 del, trisomy 18
Wilms’ tumor
13q14 del
Retinoblastoma
Osteosarcoma
Trisomy 21 (Down syndrome)
Leukemia
Monosomy 7
Leukemia and myelodysplasia syndrome
t(11q23), t(1;22), t(9;11)
Leukemia
t(1:22) (p13;q13)
Leukemia
Gonadal dysgenesis (46,XY; 45X/46,XY)
Gonadoblastoma, Germinoma
Klinefelter síndrome (XXY)
Teratoma
t(11;22)(q24;q12) t(21;22)
Ewing sarcoma/PNET
t(11;22)(p13;q12)
Intra-abdominal desmoplastic small round cell tumor
t(2;13)
Alveolar rhabdomyosarcoma
del 1p
Astrocytoma
The number of chromosomal rearrangements and gene mutations associated with tumors is now so great that almost every cancer patient undergoes some sort of molecular or cytogenetic testing. The same progression from chromosome anomaly to causative breakpoint to cancer/tumor suppressor gene (Table 15.2) can now be followed for most developmental anomalies, and epigenesis is a central factor in both neoplasia and development [20, 21].
Table 15.2
Tumor suppressor genes involved in human neoplasms
Gene | Chromosomal location | Neoplasms associated with somatic mutations | Neoplasms associated with inherited (germ-line) mutations |
|---|---|---|---|
WT-1 | 11p13 | Wilms’ tumor | Wilms’ tumor |
p53 | 17p13.1 | Most human cancers | Li-Fraumeni syndrome: carcinomas of breast and adrenal cortex; sarcomas; leukemias; brain tumors |
APC | 5q21 | Carcinomas of colon, stomach and pancreas | Familial adenomatous polyposis coli; carcinomas of colon |
DCC | 18q21 | Carcinomas of colon and stomach | Unknown |
VHL | 3p25 | Renal cell carcinoma | von Hippel-Lindau disease: retinal and cerebellar hemangioblastomas; renal cell carcinomas; angiomas and cysts of many visceral organs |
NF-1 | 17q11 | Schwannomas | Neurofibromatosis type 1; neural tumors |
NF-2 | 22q12 | Schwannomas and meningiomas | Neurofibromatosis type 2; central (acoustic) schwannomas; meningiomas |
Rb | 13q14 | Retinoblastoma; osteo- sarcoma; carcinomas of breast, bladder, prostate, and lung | Retinoblastoma; osteosarcoma |
Chromosome analysis is now merging with DNA chips to provide telomere or array analyses, capable of defining subtle deletion/duplication of any chromosome segment by its altered fluorescent pattern.
15.5 Controlling Gene Expression
Increasing gene expression occurs by increasing the number of DNA copies of that gene (gene amplification). Human folate-resistant cell cultures are seen in aggressive neuroblastomas that amplify N-Myc. The new controlling mechanisms offer novel means for understanding and manipulating developmental gene regulation. The relevance of new, non-classical pathways that control gene expression is reinforced by progressive DNA expansions/contractions and a myriad of possible gene rearrangements characteristic of most cancers.
15.6 DNA Methylation
Methylation of DNA contributes to cell differentiation, since the changes in DNA structure and expression will be transmitted to all daughter cells of a progenitor cell. Methylation of promoters probably plays a major role in cellular differentiation during embryogenesis: cells eliminate the transcription of unwanted genes by methylating their promoters.
Methylation inhibits (and occasionally activates) a very large number of genes, including some proto-oncogenes [22]. In cancers, there is often a decreased expression of DNA methyltransferase which leads to a nonspecific demethylation of 5-methylcytosine. This demethylation can reactivate the expression of proto-oncogenes, which thus act as unregulated growth-promoting agents (e.g., oncogenes, telomerases). Reactivated expression of genes involved in cell migration may lead to the emergence of more aggressive clones (clonal evolution). Abnormal methylation (hence inactivation) of anti-oncogenes similarly plays a major role in cancer development.
15.7 Zinc-Fingers
Transcription factors are those with a zinc-finger motif; zinc molecules cause these proteins to have a tertiary structure resembling fingers. The “fingers” bind the promoters of the genes under their control, to activate or inhibit them. Zinc finger proteins have multiple domains, allowing them to coordinate multiple different expression pathways. WT-1 (the Wilms’ tumor-1 gene product), the fly developmental gene hunchback, and certain hormone and retinoic acid receptors belong to the zinc-finger class of transcription factors.
15.8 Loss of Imprinting and Cancer
Genomic imprinting was discovered in the 1980s and is now recognized to be very important clinically [23–25]. Imprinting is characterized by the differential activation of alleles according to their parental origin, associated with different patterns of DNA methylation.
Mammals are diploid organisms, meaning that all somatic cells possess two copies of the genome. Each autosomal gene is therefore represented by two copies, or alleles, with one copy inherited from each parent at fertilization. For the vast majority of autosomal genes, expression may occur from either allele. However, a small proportion (<1 %) of genes are imprinted, meaning that expression occurs from only one allele. Imprinting, therefore, is defined as the parental allele-specific expression of a very limited set of genes. This is an epigenetic phenomenon whereby the DNA of the two alleles of a gene is differentially modified so that only one parental allele is normally expressed [26]. The expressed allele is dependent on its parental origin. For example, the gene encoding insulin-like growth factor II (IGF2) is only expressed from the allele inherited from the father. Similarly, human triploidy with two paternal genomes produces fetal and placental tissue (hydatidiform mole), whereas triploidy with two maternal genomes biases toward fetal tissues only (ovarian teratomas). This regulation on an epigenetic marking of parental alleles occurs during gametogenesis. Mis-regulation of imprinted gene expression or loss of imprinting refers to loss of monoallelic gene regulation and concomitant biallelic expression.
In Beckwith-Wiedemann syndrome (BWS), partial moles, complete moles, and many cancers, this equilibrium is altered, leading to loss of imprinting and IGF2 over-expression. This has been documented in Wilms’ tumor, Ewing sarcoma, rhabdomyosarcoma, adrenocortical tumors, hepatoblastoma, hepatocellular carcinoma, and pheochromocytoma. Conversely, the incidence of these tumors is greatly increased in BWS, and placentas of BWS fetuses can show mole-like changes. IGF2 is also overexpressed in choriocarcinoma, leukemias, and germ cell tumors, as well as in bladder, breast, cervical, esophageal, gastric, colorectal, pulmonary, ovarian, prostatic, renal cell and other carcinomas and tumors.
15.9 Intercellular Signaling in Development: Sonic Hedgehog
Embryonic cellular signaling pathways can be extremely complex, with core molecules that influence the development of multiple organs. A prototype is the sonic hedgehog (SHH; a member of the hedgehog gene family). In the absence of SHH protein, the Patch (PTCH) plasma membrane protein inhibits smoothened (SMO) protein. Binding of SHH with PTCH lifts this inhibition, allowing SMO to activate the SHH cascade, including GL12, GL13, CBP, and SUFU molecules [27, 28]. Mutations involving this cascade can result in a wide range of diseases, including tremors, holoprosencephaly, Greig cephalopolysyndactyly, Pallister-Hall syndrome, Rubinstein-Taybi syndrome, the nevoid basal cell carcinoma syndrome, basal cell carcinoma (syndromic and sporadic forms), medulloblastoma, meningiomas, primitive neuroectodermal tumors, breast adenocarcinomas, trichoepitheliomas, squamous cell carcinomas, esophageal carcinomas, fetal rhabdomyomas, and rhabdomyosarcomas [27].
15.9.1 Examples: Sequential Gene Expression in Growth and Neoplasia
Molecular analysis has defined many growth-related molecules through their alteration in tumors [27, 29–35]. The definition of molecular changes that fulfill Knudson’s two-hit or two-stage hypothesis has also been reviewed [36]. Although Knudson’s explanation involved one abnormal RB1 allele from the germline (predisposition or first hit), followed by somatic RB1 gene mutations in susceptible tissue (second hit in retina), epigenetic changes can also be placed on this pathway to neoplasia. This is reflected in the fact that most germline RB1 mutations originate on the paternal chromosome, implying a role for genomic imprinting/DNA methylation. Characterization of the RB1 gene as a cell cycle regulatory element places it within cell proliferation/cell death pathways.
15.10 Handling of Pediatric Tumor Specimens
Many pediatric tumors require immunostaining, molecular investigation, and electron microscopy for a definitive diagnosis and prognosis. All diagnostic pediatric tumor specimens should be submitted to the laboratory as fresh tissue immediately following biopsy when possible. The specimen should be divided into samples for formalin fixation and paraffin embedding, glutaraldehyde fixation for electron microscopy, snap freezing or storage in RNA protection medium for molecular studies such as reverse transcription-polymerase chain reaction (RT-PCR), with imprints (“dabs”) fixed for fluorescent in situ hybridization (FISH) in methanol, cytogenetic studies, or frozen for storage and subsequent analysis if required. Many molecular techniques such as FISH and RT-PCR may also be performed on routinely fixed paraffin-embedded material.
15.10.1 Wilms’ Tumor (Nephroblastoma)
Wilms’ tumor (Table 15.3) is a malignant embryonal neoplasm derived from nephrogenic blastemal cells. Several lines of differentiation are commonly seen and often replicate the histology of developing kidneys. Approximately 10 % of patients with Wilms’ tumor also have congenital abnormalities and malformation syndromes. The most common malformations are hemihypertrophy and genitourinary anomalies. The common syndromes associated with Wilms’ tumor include BWS, WAGR syndrome (Wilms’ tumor, aniridia, genitourinary abnormalities and mental retardation), and Denys-Drash syndrome (mesangial sclerosis, pseudohermaphroditism and nephroblastoma) [37–41]. Abnormalities involving the Wilms’ tumor locus, 11p13, are consistently found in the tumors of patients with WAGR and Denys-Drash syndrome. The 11p13 Wilms’ tumor locus encodes two coordinately regulated zinc-finger transcripts, WT-1 and WIT-1. These genes are highly expressed in the developing urogenital system [42–44]. The WT-1 protein binds to several sites on promoters of an IGF2 gene as well as to a promoter of the platelet-derived growth factor (PDGF) A-chain gene [45, 46]. The gene controls mesenchymal-epithelial transition during renal development. Furthermore, WT-1 expression induces transcription of one of the seven proapoptotic genes, Bak, and blocks cellular proliferation and DNA synthesis [47]. Interference with these normal regulatory influences of WT-1 may be an important factor in the genesis of nephroblastoma, which typically express high levels of IGF2 [48] and may also overproduce PDGF [49]. The expression patterns of WT-1 and Pax-2 in the metanephros overlap to a considerable extent; however, expression of WT-1 peaks as that of Pax-2 is decreasing. Therefore, it is possible that WT-1 represses Pax-2, and Pax-2 expression fails to down-regulate in the epithelial component of Wilms’ tumor and in nephroblastomatosis, the putative precursor of nephroblastoma [50]. Despite the strong association of WT1 mutations with Wilms’ tumor predisposition, WT-1 is mutated in only a minority of sporadic Wilms’ tumors [51]. This low prevalence of WT-1 abnormalities in sporadic Wilms’ tumor led to the recognition of other genes involved in pathogenesis. Evidence supporting this is provided by the linkage of familial BWS to a locus at chromosome 11p15, designated WT-2 [52, 53]. Approximately 1 % of the patients with Wilms’ tumor have a positive family history for the same neoplasm. Most of the pedigrees suggest autosomal dominant transmission with variable penetrance and expressivity. This suggests that genetic loci other than WT-1 and WT-2 are responsible for the pathogenesis of many familial as well as sporadic Wilms’ tumors [54, 55].
Table 15.3
Staging system for Wilms’ tumor cancer oncology group
Stage I | Limited to kidney and completely resected, renal capsule intact |
Fine needles aspiration or percutaneous core needle biopsy does not upstage the tumor | |
The presence of necrotic tumor or chemotherapy-induced change in the renal sinus and/or within perirenal fat should not be regarded as a reason for upstaging a tumor providing it is completely excised and does not reach the resection margins | |
Stage II | Tumor infiltrates beyond kidney but is completely resected |
(a) The tumor extends beyond the kidney or penetrates through the renal capsule and/or fibrous pseudocapsule into perirenal fat but is completely resected (resection margins clear) | |
(b) Tumor infiltrates the renal sinus and/or blood and/or lymphatic vessels outside the renal parenchyma but is completely resected | |
(c) Tumor infiltrates adjacent organs or ureter but is completely resected | |
Stage III | Gross or microscopic residual tumor confined to abdomen |
(a) Incomplete excision of the tumor which extends beyond resection margins (gross or microscopic tumor remains postoperatively) | |
(b) Any abdominal lymph nodes are involved | |
(c) Tumor rupture before or intraoperatively | |
(d) The tumor has penetrated through the peritoneal surface | |
(e) Tumor implants are found on the peritoneal surface | |
(f) Tumor thrombi present at resection margins of vessel or ureter transected or removed piecemeal by surgeon | |
(g) The tumor has been surgically biopsied (wedge biopsy) prior to preoperative chemotherapy or surgery | |
Stage IV | Hematogenous metastasis or lymph node metastasis outside the abdominal and pelvic region |
Stage V | Bilateral renal tumors at diagnosis. Each side should be sub-staged according to the above classification |
In gross examination, the most important factors to document are capsular invasion and hilar and sinus involvement along with adequate sampling of the tumor. Histology blocks should include at least one block per centimeter of the largest tumor diameter, tumor-kidney interface, capsule, hilar area, macroscopically normal kidney (at least two), and other areas of pathological significance. Some cases may be markedly cystic; in these, the tumor must be carefully examined for the presence of nodular solid areas. Particular attention should be paid to assessment of the capsule, hilar vessels, including the cut end, and renal sinus.
A triphasic Wilms’ tumor includes epithelial, stromal, and blastemal elements microscopically. Relative proportions of the epithelial components may vary widely such that one is markedly dominant (Fig. 15.1). The stromal component may vary widely from areas of stellate cells within a myxoid background to spindle cells recapitulating primitive mesenchyme. Smooth and striated muscle are the most common differentiated stromal cell types. Occasionally, a wide range of other stromal components may be present, including cartilage, osteoid, and even glial tissues.
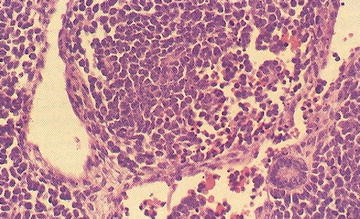
Post-chemotherapy features include necrosis, fibrosis, and foamy and hemosiderin-laden macrophages. Rapidly dividing cells, such as those of the blastemal or primitive epithelial components, tend to respond to chemotherapy, whereas the stromal elements will not. Thus, tumors with significant stromal differentiation may not reduce much in size during chemotherapy. Also, stromal components may appear to be more prominent in a post-chemotherapy specimen. The tumor that remains predominantly viable following chemotherapy and in which the main component is blastema is associated with an adverse prognosis.
15.10.2 Unfavorable Histology in Wilms’ Tumor (Anaplasia)
Nuclear anaplasia in the context of Wilms’ tumor has very specific criteria required for its definition, such as nuclear hyperchromasia, nuclear enlargement in which affected nuclei are at least three times the size of adjacent non-anaplastic nuclei, and abnormal multipolar mitoses. Using such criteria, anaplasia is present in approximately 5–10 % of Wilms’ tumors, occurring more commonly in those diagnosed in later childhood and in black patients. Anaplasia may either be focal or diffuse. Focally, one or a few, well-circumscribed, localized regions of anaplasia are found within the primary tumor that is completely excised, with the majority of tumor containing no significant nuclear atypia. Diffuse anaplasia occurs in multiple fields or outside the primary tumor and is associated with a poor response to chemotherapy. Anaplasia is of major clinical significance when present in tumors, which are potentially incompletely excised. Focal anaplasia or anaplasia in stage I cases may be of importance. Anaplasia is identified as an adverse prognostic factor. Transcription profiling indicates that a cluster of genes separates anaplastic from non-anaplastic Wilms’ tumor.
Immunohistochemical staining is often extremely useful in the diagnosis of Wilms’ tumor from a needle core biopsy. A core biopsy may simply demonstrate small round blue cells, which could represent the blastema of Wilms’ tumor or several other embryonal tumors of infancy and childhood. The most useful immunohistochemical marker is WT-1. Areas of stromal differentiation, and well-differentiated epithelial structures, may not express nuclear WT-1. Cytokeratin staining may help with the identification or confirmation of epithelial differentiation. CD56 is also useful, since primitive renal blastema is diffusely and strongly CD56 positive. Nuclear p53 is expressed in 75 % of anaplastic nephroblastomas. At present, there are no diagnostic or prognostic markers useful for the evaluation of Wilms’ tumor.
15.10.3 Nephrogenic Rests and Nephroblastomatosis
Nephrogenic rests are classically divided into perilobar and intralobar types. Perilobar rests are more common and are found in subcapsular peripheral locations with well-demarcated margins on low power. Intralobar rests present anywhere within the kidney and often have a more irregular and intermixed margin. The clinical relevance is their potential increased risk for subsequent development of Wilms’ tumor. Wilms’ tumors in association with intralobar rests develop at a younger age on average. The risk appears highest for intralobar rests in patients under 1 year of age. When a Wilms’ tumor develops within a nephrogenic rest, there is a superimposed nodular growth of the proliferating component and formation of a fibrous pseudocapsule around the tumor with the compressed rest at the periphery.
15.10.4 Cystic Nephroma and Cystic Partially Differentiated Nephroblastoma
Cystic nephroma (CN) is predominantly seen in children under 5 years of age. Its characteristic imaging appearance is of a well-circumscribed mass distinct from the surrounding kidney and predominantly cystic with thin septa. In CN, the septa contain no solid nodules and only mature elements. In cystic partially differentiated nephroblastoma, the septa also contain embryonal elements that do not form solid nodules, thus conforming to the outlines of the septa. If classical components of Wilms’ tumor are present, forming nodular structures that distort the septal contours, the lesion should be classified as cystic Wilms’ tumor. Both CN and cystic partially differentiated nephroblastoma are adequately treated by resection alone with an excellent prognosis and no chemotherapy required. Predominantly, cystic Wilms’ tumor, although almost always stage I and therefore with a correspondingly good prognosis, should be treated as an appropriately staged Wilms’ tumor. There is an association between CN and pleuropulmonary blastoma [57].
The classical pattern of CN demonstrates nests of ovoid cells separated by fibrovascular septa. Cord cells may be ovoid, epithelioid, or spindled with bland nuclei without prominent nucleoli. Septa demonstrate a marked regularly branching “chicken wire” pattern of capillaries. A wide variety of other patterns have been described. These include myxoid, sclerosing, cellular epithelioid, palisaded, spindled, storiform, and anaplastic variants. Most cases are composed of a mixture of such patterns.
15.10.5 Clear Cell Carcinoma of the Kidney
Clear cell carcinoma of the kidney comprises 3 % of malignant pediatric renal tumors. The average age at diagnosis is 3 years. There are no specific diagnostically or prognostically useful genetic features. Metastatic disease is present in 5 % of cases and up to 60 % of those have bone metastases. The tumor is solid, cystic, and cream to tan in color. Immunohistochemical stains vimentin and bcl2 are usually positive. Cytokeratin, WT1, and CD99 staining is negative [57]. The vimentin staining is unlike the dot-like pattern characteristic of renal rhabdoid tumors.
15.10.6 Renal Rhabdoid Tumor
Rhabdoid tumors are rare, comprising 2 % of pediatric renal malignancies. These tumors are predominantly shown in young children, with average age at diagnosis of 1 year. This tumor is exceptionally rare in children over 5 years of age.
In patients with renal rhabdoid tumors, genetic abnormalities of the hsNF5/INI1 tumor suppressor gene on chromosome 22 are characteristic. The gene is important for chromatin remodeling, and inactivation is tumorigenic. Mutations, deletions, and chromosome losses inactivating INI1 vary widely; therefore, a single readily available RT-PCR-based assay is not available.
Patients with renal rhabdoid tumors have an increased risk of malignant embryonal tumors of the posterior fossa; approximately 10–20 % develop a posterior fossa tumor. Rhabdoid tumors are aggressive, and the majority of patients (up to 80 %) present with metastatic disease. Grossly, rhabdoid tumors have large, solid and cystic areas with extensive areas of hemorrhage and necrosis. They are poorly defined and locally infiltrative. Microscopically, rhabdoid tumors contain eccentric nuclei and eosinophilic intracytoplasmic inclusions (Fig. 15.2). Vimentin immunostaining demonstrates characteristic strong dot-like perinuclear cytoplasmic staining in a significant proportion of cells. INI1 immunostaining is characteristically absent in the nucleus of rhabdoid tumor cells. INI1 nuclear staining is positive in the nucleus of almost all other pediatric tumors, although, renal medullary carcinoma and some cells of synovial sarcoma may also be INI1 negative.
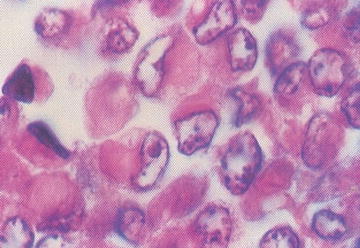
Fig. 15.2
Rhabdoid tumor cells with variable presence of intracytoplasmic inclusions (From Gilbert-Barness [56])
15.10.7 Congenital Mesoblastic Nephroma
Congenital mesoblastic nephroma is a low-grade spindle cell neoplasm affecting the renal medulla. Representing 2–5 % of pediatric tumors, congenital mesoblastic nephroma is also the most common congenital renal neoplasm. A vast majority of cases present in the first year of life (90 %). Some patients may develop hypertension. Congenital mesoblastic nephroma demonstrates a t(12;15)(p13;q25) ETV6-NTRK3 translocation, also found in congenital/infantile fibrosarcoma and secretory carcinoma of the breast. RT-PCR may be useful diagnostically [58]. Overall, 95 % of patients have an event-free survival; however, prognosis is worse with stage III with cellular congenital mesoblastic nephroma in patients 3 months or older. Grossly, the tumor has a cystic and solid whorled fibrous appearance resembling a fibroid. The cellular subtype may appear softer and more hemorrhagic.
Microscopic subtypes are classical, cellular, and mixtures and may be identified on the basis of molecular characterization and morphological features. The cellular subtype represents 40–60 % of cases and is morphologically indistinguishable from an infantile fibrosarcoma. There is increased cellularity with mitoses and focal necrosis. Mixed subtypes comprise less than 10 % of cases. The presence of incomplete excision is the main cause of local recurrence. The majority of cases with recurrence have been the cellular subtype with an abnormal gene fusion product present.
15.10.8 Renal Cell Carcinoma in Childhood
Renal cell carcinoma is uncommon in childhood and comprises 2–5 % of pediatric renal tumors. It usually occurs in children 10–15 years of age. Genetic changes are similar to adult cases. A range of familial and syndromic conditions may be associated with renal cell carcinoma development such as Von Hippel-Lindau (VHL) syndrome, hereditary papillary renal carcinoma syndrome, hereditary leiomyomatosis and renal cell cancer, Birt-Hogg-Dubé syndrome, and tuberous sclerosis. Twenty percent have metastases at diagnosis. The tumor may have a predominant clear cell, cystic papillary, or chromophobe pattern. Renal cell carcinoma is cytokeratin positive in all tumors and CD10 positive in greater than 90 %. EMA, vimentin, and CEA staining may also be positive.
15.10.9 Neuroblastoma
The incidence of neuroblastoma is 1 per 100,000 [59]. Seventy percent of patients have metastatic disease at presentation [60]. Age, clinical stage, molecular findings, and an undifferentiated subtype all influence overall survival [61]. The International Neuroblastoma Pathology Classification system [62] is as follows:
1.
Neuroblastoma, undifferentiated: No ganglion cell differentiation of neuroblasts, no definite neurofibrillary stroma on light microscopy, sheets of small round to ovoid cells.
2.
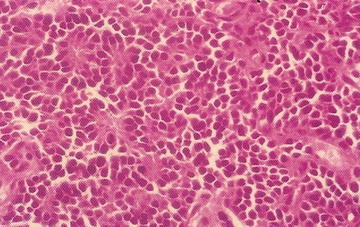
3.
Neuroblastoma, differentiating: > 5 % of neuroblasts show ganglion cell differentiation, nuclei have a prominent nucleolus. The cell has abundant eosinophilic/amphophilic cytoplasm, and the cell diameter must be more than twice nuclear diameter.
4.
Ganglioneuroblastoma, intermixed: Well-defined nests of neuroblasts within neurofibrillary background stroma that are scattered throughout the lesion. Usually presents differentiating neuroblasts within the nests of cells (Fig. 15.4). No discreet identifiable nodules are present. Less than 50 % of the lesion is represented by neuroblastoma cells. Greater than 50 % of the lesion is represented by Schwannian stroma and mature ganglion cells.
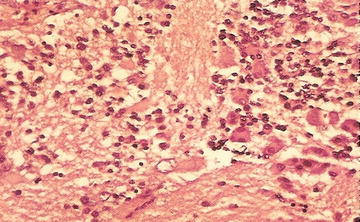
Fig. 15.4
Neuroblasts and variable ganglion cell differentiation in ganglioneuroblastoma (From Gilbert-Barness [63])
5.
Ganglioneuroblastoma, nodular: There is a macroscopic, hemorrhagic nodule within a larger bland lesion. The surrounding areas are stroma-rich or stroma dominant areas with mature or maturing ganglion cells; the well-demarcated nodule has Schwannian stroma, poor neuroblastomatous tissue; may be multiple, nodules morphologically those of neuroblastoma. Outcome of ganglioneuroblastoma, nodular is based on prognostic features of the neuroblastoma component [64], including all molecular/biological markers.
Molecular studies are mandatory for risk assessment and management in neuroblastic tumors, and it is important for optimal specimen handling to allow FISH and other molecular studies. Current biological markers associated with adverse prognosis are N-MYC amplification, DNA diploidy, 1p deletion and 17q gain. Mitosis karyorrhexis index is calculated per 5,000 neuroblasts, usually within 6–8 high-power fields in cellular tumors in a 3 μm-thick evenly stained section. The mitosis karyorrhexis index is not carried out on small needle biopsy specimens or post-treatment specimens.
Specimens are assigned to 1 of 3 prognostic categories (see Table 15.4). On immunohistochemical staining CD56 is positive and also highlights neurofibrillary stroma. NB84 is positive. CD44 is positive and associated with a better prognosis. Schwannian stroma is S100 positive.
Table 15.4
Neuroblastoma prognostic categories
Mitosis karyorrhexis index (MKI) | |
Low MKI | Fewer than 100 mitotic and karyorrhectic cells/5,000 tumor cells, or less than 2 % of tumor consisting of mitotic and karyorrhectic cells |
Intermediate MKI | 100–200 mitotic and karyorrhectic cells/5,000 tumor cells, or 2–4 % of tumor consisting of mitotic and karyorrhectic cells |
High MKI | More than 200 mitotic and karyorrhectic cells/5,000 tumor cells, or greater than 4 % of tumor consisting of mitotic and karyorrhectic cells |
International neuroblastoma staging system | |
Stage 1 | Localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes negative for tumor microscopically (nodes attached to and removed with the primary tumor may be positive) |
Stage 2A | Localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes negative for tumor microscopically |
Stage 2B | Localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes positive for tumor; enlarged contralateral lymph nodes must be negative microscopically |
Stage 3 | Unresectable unilateral tumor infiltrating across the midline, with or without regional lymph node involvement or localized unilateral tumor with contralateral regional lymph node involvement; or localized unilateral tumor with contralateral regional lymph node involvement, or midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement. The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column |
Stage 4 | Any primary tumor with dissemination to distant lymph nodes, bone marrow, liver, skin, and/or other organs (except as defined for stage 4A) |
Stage 4S | Localized primary tumor (as defined for stage 1, 2A, or 2B), with dissemination limited to skin, liver, and/or bone marrow (limited to infants less than 1 year of age). Marrow involvement should be minimal (less than 10 % of total nucleated cells identified as malignant by bone biopsy or by bone marrow aspirate). More extensive bone marrow involvement would be considered stage 4 disease. The results of the MBG scan (if performed) [57] should be negative for disease in the bone marrow |
15.10.10 Rhabdomyosarcoma
This is a malignant mesenchymal tumor with skeletal muscle differentiation. In childhood, there are two main types: embryonal (ERMS) and alveolar (ARMS). The pleomorphic type affects adults.
15.10.10.1 Embryonal Rhabdomyosarcoma
This is the most common pediatric soft tissue sarcoma. Most develop in children less than 10 years old, 50 % in those less than 5 years old, and 5 % in those less than 1 year old. 11p15 loss is reported; however, it is not diagnostically useful, and there is no characteristic fusion transcript. Despite skeletal muscle differentiation, only 5 % arise in the skeletal muscle of extremities. Fifty percent develop in the head and neck, whereas 30 % are genitourinary including paratesticular. The remainder are from a wide range of sites including bile ducts and retroperitoneum.
Microscopically there are sheets of immature round to ovoid to spindle mesenchymal cells (rhabdomyoblasts). Cellularity is variable within a loose stroma. The degree of morphological skeletal muscle differentiation varies and includes strap cells, tadpole cells, and spider cells (Fig. 15.5). Differentiation is especially prominent post-chemotherapy. The spindle cell variant consists predominantly of spindle cells with fascicles, whorls, or a storiform pattern. The Botryoid variant consists of ERMS affecting the wall beneath an epithelial surface in vagina, bladder, or bile ducts. There is an intraluminal polypoid growth pattern macroscopically. The microscopic cambium layer is a layer of neoplastic cells immediately beneath the epithelium in loose myxoid stroma. The anaplastic/pleomorphic variant has nuclear enlargement, hyperchromasia, and anaplasia.
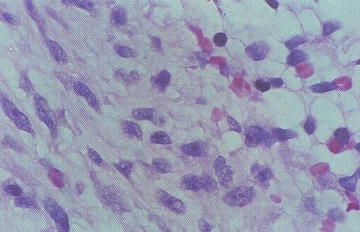
Fig. 15.5
Atypical cells with skeletal muscle differentiation in rhabdomyosarcoma (From Gilbert-Barness [56])
Immunohistochemical staining with vimentin is positive. Desmin positivity is cytoplasmic, whereas myogenin positivity is nuclear. All rhabdomyosarcomas show myogenin expression with more widespread expression in ARMS (usually greater than 70 % positive) than in ERMS (usually less than 30 % positive). Myogenin may be very focal in a limited sample such as core biopsy. MyoD1 is positive in nuclei in cases with myogenin staining. Myoglobin is positive in well-differentiated cells. Other markers may highlight aberrant expression.
15.10.10.2 Alveolar Rhabdomyosarcoma
Clinically, this tumor is the more aggressive variant of rhabdomyosarcoma. Alveolar architecture is not always present. The characteristic gene fusion transcript is present in the majority of cases, detectable by molecular techniques including FISH with FKHR break-apart probes and RT-PCR for fusion transcript deletion. Approximately 60 % have the PAX3-FKHR translocation, 20 % have PAX7-FKHR translocation, and 20 % are fusion-negative [57]. Clinical features include a rapidly growing mass with almost any site involved, and 40 % are located in the extremities. ARMS may present with disseminated disease with or without an identifiable primary site. Bone marrow metastases occur. The classic alveolar pattern consists of fibrovascular septa, tumor cell nests that may be dyscohesive (Fig. 15.6). The solid variant has sheets of round tumor cells and no septae surrounding or stroma formation. The histological features show a limited correlation with PAX/FKHR fusion status. Only totally solid alveolar architecture is associated with absence of PAX/FKHR fusion products. No morphological features predict any particular fusion type.
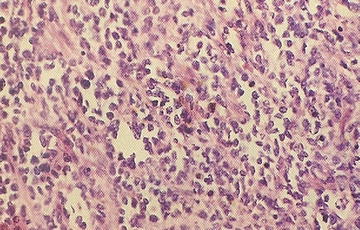
Fig. 15.6
Dyscohesion of cells resulting in the characteristic alveolar growth pattern of alveolar rhabdomyosarcoma (From Gilbert-Barness [56])
15.10.11 Hepatoblastoma
Hepatoblastoma is the most common hepatic malignancy of childhood and comprises 1 % of pediatric malignancies. Ninety percent occur in the first 5 years of life and less than 5 % are congenital. Fifty-fold have increased risk of birth weight of less than 1,000 g. There are rare predisposing syndromes in 5 % of cases such as BWS, trisomies, and familial adenomatous polyposis (FAP). Ten percent of sporadic hepatoblastomas have APC gene mutations.
In 80 % of cases, there is a single mass, whereas 20 % have multiple lesions. Anemia is present in 70 % and thrombocytosis in 50 %. Greater than 90 % of cases show elevated serum AFP concentration. Forty to 60 % are unresectable at diagnosis, but 85 % are resectable post-chemotherapy. Ten to 20 % of cases have lung metastases at diagnosis. The overall survival rate is 70 %.
Histopathologically, the tumor has a mixture of features that may be highly variable in both fetal and embryonal epithelial cells, stroma of connective tissue, osteoid, skeletal muscle or rare other epithelial types as well as teratoid features. Extramedullary hematopoiesis may be present. Immunohistochemical staining is especially useful other than to exclude other tumors.
15.10.12 Desmoplastic Small Round Cell Tumor
Desmoplastic small round cell tumor affects children and young adults, with more males affected than females. Characteristic and diagnostic is the t(11;22)(p13;q12), Ewing sarcoma (EWS)-WT1 fusion. The patient usually presents with disseminated abdominal disease. Presentation depends on the site; if aggressive, the patient has a poor prognosis. Histopathological features include infiltrative growth with desmoplastic spindle cell stroma that is variably vascular. Tumor cells are uniform, ovoid, and hyperchromatic with scant cytoplasm. Intracytoplasmic inclusions may be present. The cells are arranged in nests of variable shapes and sizes. Mitoses and necrosis may be present.
The tumor has polyphenotypic expression with CK, EMA, desmin, vimentin, and NSE staining. Anticarboxyterminus WTI is nuclear positive, and myogenin and MyoD1 are negative. Molecular techniques are required for confirmation of diagnosis.
15.10.13 Extrarenal Malignant Rhabdoid Tumor
This tumor occurs in infants and children. Congenital tumors are reported. There is an association with family history of a range of malignancies and posterior fossa tumors. Like renal rhabdoid tumors, there are abnormalities of the hSNF5/INI1 gene on chromosome 22 [57]. Clinical features consist of mass lesions at almost any site. The tumor is aggressive and has a poor prognosis; however, this has improved with current chemotherapy regimes. Histopathological features include sheets of malignant cells with vesicular nuclei, prominent nucleoli, eosinophilic cytoplasm, mitoses, and necrosis. There is a variable extent of intracytoplasmic inclusions, which are often easier to identify at the periphery or in myxoid areas. The small cell variant has cells with scant cytoplasm. Some tumors may have a markedly myxoid stroma. On immunohistochemical staining, there is co-expression of CK and vimentin. Perinuclear “dot-like” expression highlights inclusions that are composed of intermediate filaments by electron microscopy. There is variable non-diagnostic expression of other antibodies. Negative INI1 nuclear expression is essentially diagnostic.
15.10.14 Ewing Sarcoma/Primitive Neuroectodermal Tumor
EWS and primitive neuroectodermal tumor (PNET) are primary malignant small round cell tumors of bone and soft tissue. Because they have a similar phenotype and share an identical chromosome translocation, they are viewed as the same tumor, differing from each other only in their degree of neural differentiation. Tumors that demonstrate neural differentiation by light microscopy, immunohistochemistry, or electron microscopy have been traditionally labeled PNET, and those that are undifferentiated by these analyses are diagnosed as EWS. These tumors account for approximately 6–10 % of primary malignant bone tumors. Most patients are 10–15 years old, and approximately 80 % are younger than 20 years. Boys are affected more frequently than girls, although the disease occurs much more often in girls than boys during the first 3 years of life. There is a striking predilection for whites; blacks are rarely afflicted.
In EWS/PNET, approximately 85 % have a t(11;22)(q24;q12) translocation, 5–10 % of cases have a t(21;22)(q22;q12) translocation, and in less than 1 % of tumors, a t(7;22)(p22;q12) translocation is present. The fusion gene (EWS-FLI1) generated from the translocation plays a vital role in the molecular genesis of this neoplasm [57]. The aberrant gene acts as a dominant oncogene, and the resultant chimeric proteins function as aberrant transcription factors that stimulate cell proliferation.
EWS/PNET can arise in any portion of the skeleton but usually develops in the diaphysis or metadiaphysis of long tubular bones and the flat bones of the pelvis. In young children, the pelvis, ribs, and proximal long bones are preferentially affected. The tumor typically presents as a painful enlarging mass, and the affected site is frequently tender, warm, and swollen. Systemic findings that mimic infection include fever, elevated sedimentation rate, anemia, and leukocytosis.
Arising in the medullary cavity, EWS/PNET usually transgresses the cortex and periosteum, producing a soft tissue mass. The tumor is tan-white or fish flesh-like in appearance and frequently contains areas of hemorrhage and necrosis. The cells grow in sheets or irregular islands with an organoid growth pattern within which Homer-Wright rosettes may be present. Occasionally, tumor cells are larger, have irregular nuclear contours, and prominent nucleoli. Mitoses are easily identified, and necrosis may be extensive. Special histochemical stains demonstrate glycogen in up to 75 % of tumors. In a minority of cases dense core neurosecretory granules are seen by electron microscopy.
EWS/PNET cells express vimentin, CD99 (MIC-2), FLI-1, C-kit and one or more neural markers including NSE, Leu-7, S100, neurofilament, and keratin in approximately 20 % of cases [57]. EWS/PNET is usually treated with a combination of chemotherapy and surgery. The chemotherapy is given preoperatively, and chemotherapy-induced necrosis of 90 % or greater is considered a good response. Radiotherapy is reserved for tumors that are surgically inaccessible, inadequately excised tumors, and patient palliation. Effective chemotherapy has dramatically improved the prognosis to a 75 % 5-year survival of which at least 50 % are long-term cures.
15.10.15 Testicular and Paratesticular Tumors
Most germ cell tumors are seminomas in young adults; however, in younger children, they are usually teratomas and yolk sac tumors.
Seminomas. Seminomas are composed of sheets of uniform round to ovoid cells with minimal pleomorphism; in most cases, they are associated with lymphoid infiltrates. PLAP and CD117 are positive in almost all cases [57].
Embryonal Carcinoma . Embryonal carcinoma is composed of sheets, papillae, and cysts with poorly differentiated cells containing overlapping polygonal, vesicular nuclei and prominent nucleoli. CD30, CK and focal PLAP stains are positive.
Yolk Sac Tumor . This is the most common malignant germ cell tumor in infancy. Ninety percent are associated with elevated serum AFP levels and contain ovoid vacuolated cells with scattered hyaline globules. Yolk sac tumors are mitotically active. There are numerous growth patterns, and most cases contain many of the patterns: microcystic/reticular, macrocytic, solid, alveolar, endodermal sinus (Schiller-Duval bodies), papillary, myxomatous, polyvesicular/vitelline, hepatoid, and enteric (Fig. 15.7). They are cytokeratin and AFP focally positive.
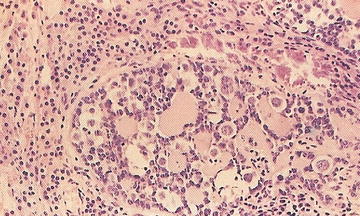
Choriocarcinoma . This tumor is very rare in childhood even as a component of mixed malignant germ cell tumor. Choriocarcinoma is positive for cytokeratin, inhibin, and hCG.
Teratoma . Teratoma occurs in infancy and is composed of tissues recapitulating all three germ layers. It is well-demarcated. Malignant areas may be present including focal PNET. It has a benign behavior in infancy.
Mixed Germ Cell Tumor . Normal immature fetal germ cells express PLAP and CD117. Infantile/congenital testicular tumors are most likely juvenile granulosa cell tumors rather than germ cell tumors.
15.10.16 Testicular Sex-Cord Stromal Tumors
Leydig Cell Tumors . This comprises less than 5 % of pediatric testicular tumors. Leydig cell tumors occur in children between ages 3 and 9 years, presenting as a painless unilateral testicular enlargement. Endocrine features may be present, including gynecomastia and precocious puberty. The tumor is a well-circumscribed, yellow-brown nodule. Histopathologically, it is composed of diffuse sheets of large polygonal cells with eosinophilic cytoplasm; Reinke crystals are present in less than 40 %. The cells have regular nuclei with prominent nucleoli. Malignant behavior occurs in 10 % of tumors, particularly tumors greater than 5 cm in diameter and those containing cytological atypia, mitoses, necrosis and vascular invasion. The cells are vimentin and inhibin positive [57].
Related posts:
 Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
Molecular-Genetic Testing in Penile, Scrotal, and Testicular Cancer
Molecular Diagnostics of Pancreatic Cancer
 Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Pancreatic Endocrine Neoplasms
Molecular Pathology and Diagnostics of Cutaneous Malignancy
Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Pancreatic Endocrine Neoplasms
Molecular Pathology and Diagnostics of Cutaneous Malignancy
 Molecular Diagnostics of Myeloid Neoplasms
Molecular Diagnostics of Myeloid Neoplasms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


