Myeloproliferative neoplasms
Chronic myelogenous leukemia, BCR-ABL1-positive
Chronic neutrophilic leukemia
Polycythemia vera
Primary myelofibrosis
Essential thrombocythemia
Chronic eosinophilic leukemia, NOS
Mastocytosis
Myeloproliferative neoplasm, unclassifiable
Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1
Myelodysplastic/myeloproliferative neoplasms
Chronic myelomonocytic leukemia
Atypical chronic myeloid leukemia, BCR-ABL1 negative
Juvenile myelomonocytic leukemia
Myelodysplastic/myeloproliferative neoplasm, unclassifiable
Myelodysplastic syndromes
Refractory cytopenia with unilineage dysplasia
Refractory anemia with ring sideroblasts
Refractory cytopenia with multilineage dysplasia
Refractory anemia with excess blasts
Myelodysplastic syndrome associated with isolated del(5q)
Myelodysplastic syndrome, unclassifiable
Childhood myelodysplastic syndrome
Acute myeloid leukemia (AML) and related precursor neoplasms
AML with recurrent genetic abnormalities
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFβ-MYH11
Acute promyelocytic leukemia with t(15;17)(q22;q21); PML-RARA
AML with t(9;11)(p22;q23); MLLT3-MLL
AML with t(6;9)(p23;q34); DEK-NUP214
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
AML with mutated NPM1
AML with mutated CEBPA
AML with myelodysplasia-related changes
Therapy-related myeloid neoplasms
Acute myeloid leukemia, not otherwise specified
AML with minimal differentiation
AML without maturation
AML with maturation
Acute myelomonocytic leukemia
Acute monoblastic and monocytic leukemia
Acute erythroid leukemia
Acute megakaryoblastic leukemia
Acute basophilic leukemia
Acute panmyelosis with myelofibrosis
Myeloid sarcoma
Myeloid proliferations related to down syndrome
Blastic plasmacytoid dendritic cell neoplasm
18.2 Diagnostic Algorithms
The morphologic examinations on peripheral blood smears, bone marrow smears and bone marrow biopsies are always the first step on diagnostic algorithms, which are followed by immunophenotyping, cytogenetics, molecular cytogenetics and molecular genetics studies. The initial morphologic evaluation of the available specimen including peripheral blood and/or bone marrow aspirate is critical for the subsequent ordering of ancillary studies.
Generally, the presence of 20 % or more blasts in bone marrow or peripheral blood warrants the diagnosis of acute leukemia. However, when certain cytogenetic changes e.g. t(8;12)(q22;q22), inversion 16/t(16;16)(p13.1;q22), t(15:17)/(q22;q12) are confirmed, the diagnosis of leukemia can be made even when the blast count is less than 20 %. Careful assessment of morphology of blast with monocytic differentiation is needed since monoblasts and promonocytes are both counted as blasts, while atypical monocytes are not. Blasts with bilobed, or cup-shape or concaved nuclei with or without fine azurophic granules and blasts with Auer rods in cytoplasm are also important to recognize, which could suggest acute promyelocytic leukemia (APL). In this situation, a laboratory work up for disseminated intravascular coagulopathy (DIC) is warranted and molecular genetic study of t(15;17)(q22;q12)/PML-RARA should be performed to confirm the diagnosis. Blasts with perinuclear hofs and chunky orange, or salmon-pink cytoplasmic granules are also important to recognize, not to be misinterpreted as myelocytes, which are unique features of acute leukemia with t(8;21)(q22;q22)/RUNX1-RUNX1T1. An overt increase of abnormal eosinophils with large, basophilic granules, along with increased myeloblasts, are features of acute leukemia with inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB-MYH11. Immunophenotyping by flow cytometry is required to define the lineage differentiation of the blast cell population. Cytogenetic studies by conventional karyotyping or FISH analysis should be performed to detect the recurrent cytogenetic abnormalities. According to studies, approximately 55 % of acute myeloid leukemia cases have various kinds of cytogenetic changes [2]. Some of the cytogenetic aberrations also predict disease prognosis. Mutation analysis of certain genes, such as NPM1, CEBPA, FLT3 internal tandem duplications (FLT3-ITDs) and FLT3 kinase domain mutation (FLT3-TKD), have been included in the 2008 WHO classification, and these mutations are not only related to the nature of AML but also tend to be considered as important prognostic markers. Moreover, clinical findings should be integrated into the diagnosis consideration, as in patients with myeloid proliferations related to Down syndrome and therapy-related myeloid neoplasms. Schematic algorithms of clinical and genetic diagnostic approaches for acute myeloid leukemia is summarized below (Fig. 18.1) (modified from http://www.uscap.org/site~/98th/pdf/companion21h02.pdf; accessed Jan 10, 2011) [3].
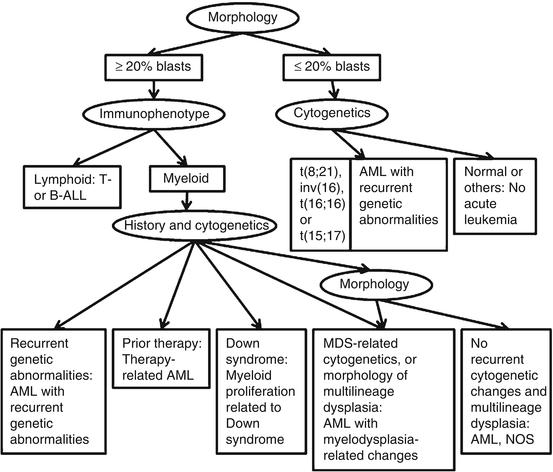
Fig. 18.1
Algorithmic approach to the classification of acute leukemia (Modified from http://www.uscap.org/site~/98th/pdf/companion21h02.pdf)
For myeloproliferative neoplasm (MPN), myelodysplastic syndromes/myeloproliferative neoplasm (MDS/MPN), and myelodysplastic syndrome (MDS), the diagnostic algorithms integrate clinical and laboratory data, histologic findings, and molecular markers. The 2008 WHO classification criteria combine all three factors [1]. Clinical and laboratory data are important. For example, in patients with hemoglobin level greater than 18.5 g/dL in men or 16.5 g/dL in women, polycythemia vera (PV) would be suspected if the patients do not have any other etiology that could cause a secondary erythrocytosis or elevated hemoglobin, and testing JAK2 mutation (V671F, or exon 12) is needed to confirm the diagnosis. A patient with essential thrombocythemia would have a platelet count of more than 450 × 109/L, persistent, often accompanying thrombotic episodes. Bone marrow morphology is an important diagnostic tool, which is non-replaceable to distinguish among the categories of MPN, MDS/MPN, and MDS, but plays only a minor role in distinguishing between reactive and clonal myeloproliferation [1]. True MPN could be distinguished from MDS and MDS/MPN by the absence of dyserythropoiesis, dysgranulopoiesis and monocytosis, and the morphology of hyperplastic megakaryocytes provides important diagnostic clues [4]. MDS will be considered when morphologic dysplasia is present in one or more major myeloid cell lineages with associated cytopenia. The availability of molecular markers, namely, BCR-ABL1, JAK2 V617F, JAK2 exon 12, MPL 515L/K, c-Kit D816V are widely applied in the diagnosis of myeloproliferative neoplasm. Among them, detection of t(9;22)(q34;q11.2)/BCR-ABL1 translocation is necessary to diagnose chronic myelogenous leukemia (CML), which makes the diagnostic criteria dramatically different from other categories of myeloid disorder. Different from the prior versions of WHO classification, novel 2008 WHO classification has conjunction with the cytogenetic aberration and certain molecular markers into major diagnostic criteria of many categories of myeloid disorders [5]. We will discuss the important diagnostic features and correlated clinicopathologic findings, and prognostic outcomes of myeloid neoplasm as below in this chapter.
18.3 Myeloproliferative Neoplasms (MPN)
The MPN are clonal hematopoietic stem cell disorders characterized by proliferation of one or more of the myeloid lineage [1]. The bone marrow is hypercellular with effective hematopoietic maturation and the peripheral blood has increased numbers of granulocytes, red blood cells, and/or platelets. This category includes eight entities: Chronic myelogenous leukemia (CML), BCR-ABL1-positive, chronic neutrophilic leukemia, polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia (CEL), NOS, mastocytosis (MS), and myeloproliferative neoplasm, unclassifiable (MPN, U).
18.3.1 BCR-ABL and Chronic Myelogenous Leukemia (CML)
CML is a clonal proliferation of myeloid cells at all stages of differentiation with the chromosome translocation t(9;22)(q34;q11.2) leading to the formation of the BCR-ABL fusion gene. Patients usually present with marked leukocytosis and basophilia in the peripheral blood and bone marrow with splenomegaly. An initial chronic phase usually lasts for 3–5 years and the disease progresses to accelerated phase and blast phase, which have increased blast counts. The blast phase has more than 20 % of blasts and it can be of myeloid or lymphoid lineage or mixed lineages.
As early as the 1960s, Philadelphia (Ph) chromosome, a minute chromosome 22 resulting from a translocation of chromosome 22q11.2 to chromosome 9q34 [t(9;22)(q34;q11.2)], was identified and has been associated with CML since 1973 [6, 7]. Reportedly 90–95 % of CML cases have this translocation at diagnosis [1], and the remaining cases have either variant translocations involving chromosome other than chromosomes 22 and 9, or cryptic translocations which cannot be identified by conventional cytogenetic analysis. All of these cases have molecular level detectable BCR-ABL1 transcript, a fusion product of BCR gene on chromosome 22 with ABL1 gene on chromosome 9 [8]. The BCR-ABL1 fusion gene is present in CML by definition, and has been designated in 2008 WHO classification as CML, BCR-ABL1 positive. However, BCR-ABL1 gene rearrangement is not specific for CML, as it is also found in acute lymphoblastic leukemia (ALL), AML, and rarely, MDS [9–12].
As known, the wild-type BCR gene encodes a serine/threonine kinase and might be involved in signal transduction pathways [13]. The wild-type ABL1 gene product is a tyrosine kinase involved in cell cycle regulation [14]. The BCR-ABL1 fusion gene produces a chimeric oncoprotein, BCR-ABL1, which is an aberrantly activated tyrosine kinase and activates several different growth factor mediated signaling pathways including RAS, STAT, NFκB, PI3K/AKT, and MAPK pathways [15].
The breakpoint in the BCR gene may occur at three different sites: major (M-BCR), minor (m-BCR), and micro (μ-BCR). M-BCR spans exons 12–16, and encodes an abnormal protein, p210. M-BCR is the most common site in CML, occurring in 95 % of CML patients. m-BCR, spanning exons 1–2, encodes a shorter protein, p190. μ-BCR occurs rarely; it spans exons 17–20 and encodes a larger fusion protein, p230 (Fig. 18.2). μ-BCR (p230) is associated with prominent neutrophilic maturation and/or conspicuous thrombocytosis [1]. m-BCR (p190) is most frequently associated with Ph chromosome positive ALL. However, p190 can be seen in rare cases of CML with monocytosis which resembles chronic myelomonocytic leukemia and a small amount of p190 can be seen in the majority of p210 CML as a result of alternative splicing [1]. A more recent study reported p190 BCR-ABL CML, although rare, is associated with a poor outcome and irresponsive to tyrosine kinase inhibitor therapy [17].
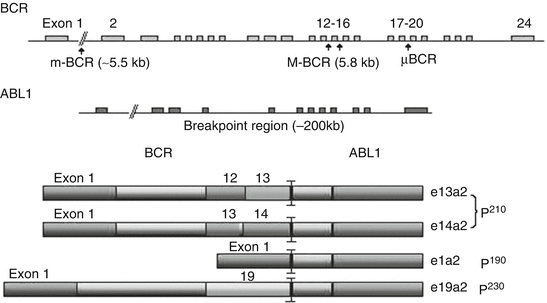
Fig. 18.2
Schematic BCR-ABL1 fusion proteins. Arrows indicate breakpoint sites for M-BCR (p210), m-BCR (p190), and μ-BCR (p230). The breakpoints in BCR are variable and can occur in the major (M-BCR), minor (m-BCR), or micro (μ-BCR) breakpoint cluster region. The breakpoint in ABL1 occurs upstream of 2a. The resulting fusion transcripts are e13a2, e14a2, e1a2, e19a2 encoding p210, p190, and p230, respectively
To diagnose CML, it needs a correlation of clinical features, laboratory findings, bone marrow morphology, and cytogenetic and molecular studies. Detection of Ph chromosome or BCR-ABL1 fusion gene is required. Conventional cytogenetics has been the gold standard, but fluorescence in-situ hybridization (FISH) and reverse transcriptase PCR (RT-PCR) methods are more sensitive for diagnosing the disease and monitoring for minimal residual disease. FISH can detect a cryptic translocation as well as deletions of derivative chromosome [18]. RT-PCR is most sensitive and real-time PCR methods can be used for quantification [19, 20].
18.3.2 CML Transformation, Therapy Monitoring and Imatinib Resistance
The accelerated and blast phases of CML frequently demonstrate secondary cytogenetic changes, including extra Ph chromosome, +8, +19, or i(17q) [1]. Translocations characteristic of de novo AML are occasionally detected in blast phase of CML, such as t(8;21)(q22;22), t(3;21)(q26;q22), t(7;11)(p15;p15), inv16(p13q22), and t(15;17)(q22;q12-21), which are, though with the same chromosome breakpoints, not associated with a better prognosis as they are noted in de novo AML. Associated gene changes include TP53, RB1, MYC, p16INK4a, RAS, AML1 and EVI-1, but their significance in diagnostics is unknown [1].
Targeted on the BCR-ABL1 tyrosine kinase activity, imatinib mesylate is very effective to control CML, particularly when used early in the chronic phase. Imatinib mesylate induces hematologic remission in nearly all CML patients [21]. The evaluation of response to treatment is based on the hematologic, cytogenetic, and molecular criteria. A hematological response (HR) is indicated by normal peripheral blood cell counts and bone marrow morphology. Complete, major, minor, and minimal cytogenetic response (CyR) can be achieved by detection of 0 %, < 35 %, 36–65 % and 66–95 % Ph chromosome positive (Ph+) bone marrow cells [21]. Molecular response (MR) is defined by reduction in the BCR-ABL1 transcript, which can be measured by quantitative RT-PCR. The real-time technology provides sensitive and reproducible results. A ratio of the BCR-ABL1 to control genes, usually ABL1, is reported, and the treatment plan is changed accordingly [22].
Resistance to imatinib mesylate occurs when subclones of CML cells acquire point mutations on ABL1 kinase domain that prevent the binding of imatinib mesylate to the kinase. Up to 90 % of the resistant patients have detectable mutations, and more than 35 mutations have been described [23]. Detection of emerging mutations can be analyzed by PCR followed by direct sequencing of the BCR-ABL1 kinase domain. However, it should be pointed out that kinase mutation independent imatinib-resistance also exists in CML, e.g. LYN kinase overexpression, and increased BCR-ABL1 expression [24, 25]. A study demonstrates a role of constitutive activation of the PI3K/AKT1 pathway in BCR-ABL1-independent imatinib resistance [26]. Additionally, some patients are resistant to imatinib primarily, which is defined by inability to achieve complete hematologic response by 3 months or molecular complete response by 6 months of imatinib treatment, and the molecular mechanism remains unclear.
18.3.3 JAK2 and MPN
In 2005, several groups independently identified JAK2 V617F as the major molecular mutation event in BCR-ABL1 negative MPNs. This somatic mutation is found in more than 95 % of PV patients, and 40–50 % of ET and PMF patients [27–30]. With this regard, PV, ET and PMF may be considered as a spectrum of diseases sharing a common molecular etiology. There is also a rare JAK2 exon 12 mutation which has similar functional effects as JAK2 V617F [1, 31].
JAK2 (Janus Kinase 2) is a tyrosine kinase that mediates activation of receptors for erythropoietin, thrombopoietin, granulocyte-macrophage colony-stimulating factor, and granulocyte colony-stimulating factor. It is also involved in signaling by receptors with tyrosine kinase activity, such as KIT [32]. JAK2 mutations can activate the downstream signaling pathways (STAT, ERK1/2, MAPK, PI3K/AKT) and cause uninhibited growth, independent of cytokines [33, 34].
Virtually all patients with PV have either JAK2 V617F or JAK2 exon 12 mutation, thus, it is a sensitive diagnostic marker for PV. Homozygous mutation of JAK2 V617F has been more frequently detected in PV and associated with the clinical picture of classic PV and with a higher tendency to secondary myelofibrosis but without increased leukemia [35]. For ET or PMF, only about half of patients have the JAK2 mutation and a small portion of patients (5 %) have MPL mutation (MPL W515K/L) [36], so the diagnostic use of JAK2 mutation is limited. JAK2 V167F is not specific to any of the MPNs and its absence does not exclude MPN; at the same time, its presence does support the diagnosis of an MPN if other clinical and laboratory data are supportive. Importantly, its presence excludes reactive myeloproliferative causes [4]. In addition, concurrent MPL and JAK2 V617F mutations have also been reported, which indicate that these two mutations may have functional complementation in myeloproliferative disease [36]. Of note as well, JAK2 mutation could occasionally be seen in MDS [37], MDS/MPN [38] and rarely coexist with CML [39].
For the diagnosis of PV, ET and PMF, an initial JAK2 V617F test is routinely performed. If the result comes back negative, a JAK2 exon12 mutation test can be done if PV is still suspected, while an MPL W515K/L mutation should be performed if ET or PMF is suspected. Either DNA or RNA from the peripheral blood or bone marrow aspirate is applicable for the mutation analysis. Qualitative as well as quantitative methods, including restricted fragment length polymorphism (RFLP), direct Sanger sequencing, and real time PCR, can be selected. Real time PCR is a preferred method with better sensitivity.
18.3.4 Polycythemia Vera (PV)
PV is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The proliferation involves not only the erythroid lineage but also the granulocytes and megakaryocytes. The diagnosis of PV is based on clinical and laboratory parameters. According to 2008 WHO classification, PV is suspected in patients with hemoglobin levels greater than 18.5 g/dL in men or 16.5 g/dL in women, or hemoglobin greater than 17 g/dL in men or 15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from a previous base line [1]. Serum erythropoietin level is usually lower than that in the other pseudo PVs and also below the normal range in healthy patients (3.3 IU/L) [40]. Bone marrow biopsy shows marked hypercellularity with pan-hyperplasia. Lack of stainable iron particles is a common phenomenon noted in marrow aspirate from PV patient. JAK2 V617F or other functionally similar mutations such as JAK2 exon 12 mutation is a required molecular test. Identification of an abnormal karyotype would be helpful but not necessary for a diagnosis since only about 20 % of patients with PV have cytogenetic abnormalities. Presence of Ph chromosome and/or BCR-ABL fusion excludes PV. The most common recurring cytogenetic abnormalities include +8, +9, del (20q), del (13q) and del (9p); sometimes +8 and +9 are found together [41].
18.3.5 Essential Thrombocythemia (ET)
ET is characterized by sustained thrombocytosis > 450 × 109/L in the peripheral blood and increased numbers of large, mature megakaryocytes in the bone marrow. Bone marrow cellularity usually retains in normal range. There is no significant erythrocytosis or leukocytosis with left shifted mutation. Clinically the patients usually have episodes of thrombosis and/or hemorrhage. The diagnosis is after exclusion of other causes of reactive or neoplastic thrombocytosis, including other MPN, inflammatory, iron deficiency anemia, infectious or non-hematopoietic neoplastic disorders. The presence of a BCR-ABL1 fusion gene excludes the diagnosis of ET. As mentioned above, approximately 40–50 % of ET patients have the JAK2 V617F or a functionally similar mutation. It has been recently found that carrying homozygous JAK2 V617F mutation in patients with ET increased a risk of recurrent thrombosis [42]. MPL mutation, namely MPL W515K/L, has been found in 1 % of cases of JAK2 V617K negative ET [1]. MPL is a thrombopoietin receptor, and it is spontaneously activated by the mutation in amino acid position 515 (MPL W515K/L). The mutation leads to thrombocytosis and myelofibrosis [36]. Worthy of mention is that W515L/K has not been detected in patients with PV according to a large series of studies including 1,182 patients with myeloproliferative and other myeloid disorders [6]. Thus, presence of W515L/K narrows the diagnosis down to ET or PMF. Presence of JAK2 or MPL mutation helps to make the diagnosis, nevertheless, neither JAK2 nor MPL mutations is specific for ET.
There is no consistent karyotypic abnormalities noted in ET, but +8, abnormalities of 9q, and del (20q) have been reported [43].
18.3.6 Primary Myelofibrosis (PMF)
PMF is characterized by a proliferation of predominantly megakaryocytes and granulocytes in the bone marrow associated with excess reticulin and collagen deposition and extramedullary hematopoiesis (EMH). In cellular phase, PMF is often associated with myeloid preponderance, with less intensive myelofibrosis besides atypical megakaryocytic proliferation. Historically, PMH should have no other myeloproliferative neoplasm diagnosed before and no BCR-ABL1 fusion gene documented. As aforementioned, approximately 50 % of patients with PMF have the JAK2 V617F mutation [27–30]. Up to 5 % of PMF cases have MPL W515K/L mutation [27–30, 36], although none of the mutations are specific to the disease entity. Similar to ET, the presence of JAK2 V617F or MPL mutation is helpful to rule out reactive marrow fibrosis, e.g. chronic inflammatory condition, autoimmune disorder, and drug-related situations. Exclusion of other primary and secondary malignancies with secondary myelofibrosis, namely lymphoma, metastatic carcinoma, is also needed.
Approximately 30 % of patients have cytogenetic changes, including partial trisomy 1q, 13q deletion, 20q deletion, trisomy 8, and chromosomes 1, 7, and 9 abnormalities [44–46]. The presence of either del(13)(q12–22) or der(6)t(1;6) (q21–23;p21.3) is strongly suggestive but not diagnostic of PMF [44, 47].
Since some cytogenetic abnormalities, e.g. 20q deletion and trisomy 8, can also be associated with MDS, differential diagnosis should include fibrotic MDS. A careful morphologic assessment and correlation with clinical presentation would be helpful. In general, splenomegaly is less commonly associated with MDS. Presence of characteristic syncytial clustering of megakaryocytes, extrasinasoidal hematopoiesis and osterosclerosis favors PMF over fibrotic MDS.
18.3.7 Mastocytosis
Mastocytosis is a new addition to the MPN category in 2008 WHO classification. It is a clonal proliferation of mast cells that accumulate in one or more organ systems, and it is characterized by the presence of multifocal compact clusters or cohesive aggregates/infiltrates of abnormal mast cells [1]. Cutaneous and systemic mastocytosis are the two most common clinicopathologic settings. Systemic mastocytosis with associated clonal hematopoetic non-mast cell lineage disease (SM-AHNMD) has been observed and considered a variant of systemic mastocytosis [1]. Mastocytosis is separated from mast cell hyperplasia by distinct morphological and/or molecular characteristics.
KIT mutations at codon 816 in exon 17, most commonly D816V, have been reported in 95 % or more of adult patients with systemic mastocytosis or one third of cutaneous mastocytosis in pediatric patients [48]. The KIT mutation results in a constitutively activated kinase, signaling from which appears to be essential for the survival and proliferation of mast cell precursors in the marrow. The D816V mutation also confers resistance to imatinib therapy [49]. Other mutations on KIT including D816Y, D816H and D816F are rarely seen. The latter (D816F) is reported and relatively more common in the pediatric group [50]. Pyrosequencing or mutation-specific quantitative PCR on DNA extracted from microdissected bone marrow or skin biopsy specimens, or from sorted mast cells from bone marrow aspirates are recommended methods [51, 52].
In the setting of SM-AHNMD, seeking additional disease specific cytogenetic or molecular abnormalities is warranted. For example, JAK2 mutation can be detected in MPN. In addition, since D816V is also observed in cases of AML, particularly in tumors with core binding factor (CBF) translocations [53, 54], correlation with clinical information and morphologic findings is suggested before a final diagnosis is rendered.
18.3.8 Chronic Neutrophilic Leukemia (CNL)
CNL is very rare, characterized by persistent neutrophilia (>80 % of white blood cells, white blood cells at least > 25 × 109/L) in peripheral blood, bone marrow hypercellularity and hepatosplenomegaly. Diagnosis is made by exclusion of other causes of neutrophilic leukocytosis. There is no Philadelphia chromosome or BCR-ABL1 fusion gene. No PDGFRA, PDGFRB, FGFR1 gene rearrangements are detected. JAK2 mutation has been found in 20 % of patients [55]. In the presence of JAK2 mutation, further investigation is requested to completely exclude any JAK2 positive MPN as well as MDS/MPN.
18.4 Myeloid and Lymphoid Neoplasms with Eosinophilia and Abnormalities of PDGFEA, PDGFB, or FGFR1
Myeloproliferative and lymphoid neoplasms associated with rearrangement of PDGFRA, PDGFRB and FGFR1 includes three rare diseases. They have some shared clinical and pathological features and all of them have chromosomal rearrangement of one of three tyrosine kinases: platelet-derived growth factor receptor (PDGFR) alpha (A), PDGFR beta (B), or fibroblast growth factor receptor 1 (FGFR1) [1]. The fusion genes encode aberrant tyrosine kinases. Eosinophilia is characteristic but not invariable. All three diseases could represent as AML, MDS/MPN or MPN, depending upon their partnership. The most common partner genes involved are FIP1L1, ETV6/TEL, and ZNF198 for PDGFRA, PDGFRB, and FGFR1, respectively [56]. Cytogenetic and molecular genetic analysis should be performed. FIP1L1-PDGFRA fusion gene usually results from a cryptic del(4)(q12). Clinically, the specific gene fusion is representative of a picture of chronic eosinophilic leukemia (CEL) with eosinophilia with dysplasia. ETV6-PDGFRB results from t(5;12)(q31 ~ 33;p12), which has hematological features resembling CMML, CEL, aCML with eosinophilia and MPN with eosinophilia [57–59]. Translocations involving an 8p11 breakpoint lead to a fusion gene with FGFR1 [1]. There are multiple partner genes associated with FGFR1. Among them, one of the partners located at 6q27 creates a fusion gene, FGFR10P1/FGFR1, is observed in some patients with PV [60]
To diagnose PDGFR or FGFR related disorder, FISH or PCR should be performed. FISH analysis with any PDGFA, PDGFRB or FGFR1 probe is widely accepted to identify the gene rearrangement. PCR is an alternative approach. Recently, generic quantitative RT-PCR has also been developed to detect diverse PDGFRA or PDGFRB [61].
18.5 Myelodysplastic/Myeloproliferative Neoplasms (MDS/MPN)
MDS/MPN neoplasms have both MDS and MPN features, comprising chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia, BCR-ABL1 negative (aCML), juvenile myelomonocytic leukemia (JMML), and MDS/MPN, unclassifiable.
The manifestation of CMML is persistent peripheral monocytosis greater than 1 × 109/L and fewer than 20 % blasts (including promonocytes). Philadelphia Chromosome/BCR-ABL1 gene or rearrangement of PDGFRA or PDGFRB should be absent. Dysplasia involves one or more myeloid lineages. Dysplasia is not a necessary condition for diagnosis of CMML if the other criteria are met. The other diagnostic criteria include a persistent monocytosis lasting over 3 months without any other explainable reactive process, or a clonal cytogenetic or molecular genetic aberration is acquired. The most common numerical and structural chromosome abnormalities associated with CMML are trisomy 8, deletion of 7 or 7q and 12p. In view of molecular changes, as many as 40 % of patients with CMML have RAS gene point mutations [62, 63].
However, the diagnosis of CMML is based on clinical, laboratory and morphological observations. Nonspecific cytogenetic changes are found in 20–40 % of patients [1]. It is important to know that cases with abnormalities of 11q23 suggest the diagnosis of AML instead of CMML. Cases with p190 BCR-ABL1 is classified as CML, regardless of persistent high monocyte count over 10 % of WBCs or 1 × 109/L [64].
Atypical chronic myelogneous leukemia (aCML) is characterized by leukocytosis with dysplastic neutrophils and their precursors. BCR-ABL1 fusion gene is absent. PDGFA or PDGFB genes are also excluded. Similar to BCR-ABL1 positive CML, patients with aCML commonly present with elevated WBC count with granulocytic predominance and splenomegaly. It is distinguished from CML by dysgranulopoiesis with nuclear aberrations and cytoplasmic hypogranulation [64, 65] and from CMML by less than 10 % monocytes [64]. About 30 % of cases have acquired mutations of NRAS or KRAS [66]. Cytogenetic abnormalities have been reported in up to 80 % of patients with aCML, most commonly +8 and del(20q).
JMML affects children of 0–14 years of age. There is no BCR-ABL1 fusion gene. PTPN11 mutations occur in 35 % of patients [67, 68], and oncogenic mutation of the RAS, NRAS, KRAS, and NF1 are each seen in approximately 20 % of patients. Most of the patients with JMML (65 %) have normal karyotype. Monosomy 7 is found in about 25 % of patients. Other cytogenetic abnormalities are also noted in 10 % of patients [1].
18.6 Myelodysplastic Syndrome (MDS)
MDS is a group of clonal hematopoietic stem cell diseases characterized by cytopenia, dysplasia in one or more of the major myeloid cell lines, ineffective hematopoiesis, and increased risk to transform to AML. According to 2008 WHO classification, the diseases include refractory cytopenias with unilineage dysplasia (RCUD), refractory anemia with ring sideroblasts (RARS), refractory cytopenia with multilineage dysplasia (RCMD), refractory anemia with excess blasts (RAEB), myelodysplastic syndrome – unclassified (MDS-U), and MDS associated with isolated del(5q). This classification integrates prognostically different subgroups, and certain chromosome aberrations, which have prognostic significance and may predict response to therapeutic drugs. For example, MDS associated with isolated del(5q), which usually has a favorable prognosis and may benefit from lenalidomide therapy [69, 70].
Cytogenetic abnormalities have been found in approximately 50 % of patients with MDS. Table 18.2 shows the common recurring chromosomal changes. The presence of these chromosomal abnormalities and refractory cytopenia of undetermined origin could suggest a diagnosis of presumptive MDS if no convincing morphologic evidence of dysplasia. Exceptions are sole +8, −Y or del(20q), which are not sufficient for a diagnosis of MDS if morphological criteria are not met. These recurring changes can also be used in monitoring of the disease during follow up. It is known that certain cytogenetic changes are associated with some characterized clinical or morphologic features. For example, del(5q) occurs primarily in elderly women with refractory macrocytic anemia, and the associated MDS is characterized by megakaryocytes with non-lobated or hypolobated nuclei, and has a favorable clinical course. Loss of 17p is associated with pseudo Pelger-Huët anomaly, small vacuolated neutrophils, TP53 mutation and unfavorable clinical course [1, 71]. Inversion of 3q21q26 or t(3;3;)(q21;q26.2) is often associated with atypical megakaryocytic hyperplasia [72]. Complex karyotype which is defined by at least three independent chromosomal abnormalities, typically includes chromosomes 5 and/or 7 [−5/del(5q), −7/del(7q)], and is associated with an unfavorable clinical course [1, 73].
Cytogenetic abnormalities | Frequency in MDS | Frequency in t-MDS |
|---|---|---|
Unbalanced | ||
+8 | 10 % | |
−7 or del(7q) | 10 % | 50 % |
−5 or del(5q) | 10 % | 40 % |
del(20q) | 5–8 % | |
−Y | 5 % | |
i(17q) or t(17p) | 3–5 % | |
−13 or del(13q) | 3 % | |
del(11q) | 3 % | |
del(12p) or t(12p) | 3 % | |
del(9q) | 1–2 % | |
idic(X)(q13) | 1–2 % | |
Balanced | ||
t(11;16)(q23;p13.3)
Related posts: Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Gynecologic Malignancies
Molecular Pathology of Bone and Soft Tissue Neoplasms and Potential Targets for Novel Therapy
Molecular Pathology and Diagnostics in Esophago-gastric Cancer
Molecular-Genetic Testing in Hepatocellular Carcinoma and Its Premalignant Conditions
Molecular Pathology and Diagnostics of Colorectal Cancer Molecular Pathology and Diagnostics of Gliomas
Molecular Pathology and Diagnostics of Gynecologic Malignancies
Molecular Pathology of Bone and Soft Tissue Neoplasms and Potential Targets for Novel Therapy
Molecular Pathology and Diagnostics in Esophago-gastric Cancer
Molecular-Genetic Testing in Hepatocellular Carcinoma and Its Premalignant Conditions
Molecular Pathology and Diagnostics of Colorectal Cancer
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||