Figure 59-1 Immunohistochemistry (A) (1) An antibody specific for a particular protein, phosphate, sulfate, carbohydrate, or other moiety is allowed to bind to the tissue section. (2) A secondary “anti-antibody” is added, bearing numerous biotin tags. (3) A complex of avidin and horseradish peroxidase (HRP) is added. Avidin binds tightly to the biotin on the secondary antibody, while HRP reacts with a chromogen in the presence of H2O2, producing a visible precipitate on the tissue. (B) Microscopic images of a gastrointestinal stromal tumor. The brown staining in the right image represents immunohistochemical detection of the KIT tyrosine kinase (CD117) in the tumor.
Although immunohistochemistry is a mainstay in modern pathology, it does have limitations. Not all antibodies are perfectly specific; background issues and cross reactivity can affect interpretation. Further, the technique is at best only semiquantitative and can be influenced by the length of formaldehyde fixation and many other parameters. Some of these limitations can be mitigated by automation of the staining, but tumor necrosis, intraoperative ischemia, and variation in specimen processing are all factors that can complicate the reading of immunohistochemical stains.
Immunofluorescence
Because formaldehyde fixation greatly distorts or even destroys many epitopes in tissue samples, there are numerous antibodies that will not work on FFPE sections. Such antibodies may still be useful, however, on cryostat sections, which can be prepared if fresh-frozen tissue is available. In these circumstances, fluorescence rather than histochemistry is the preferred method of detection, as it has greater sensitivity and allows for several antibodies to be stained at once (using different fluorophores). 9 Immunofluorescence is routinely used in evaluating medical diseases of the kidney and skin, but is rarely employed in cancer diagnosis.
In Situ Hybridization
Expression of particular mRNAs in tumor tissue can be measured by in situ hybridization (ISH). 10 In this approach, complementary oligonucleotide probes (either DNA or RNA) bearing a chemical tag such as digoxigenin are allowed to hybridize to their target mRNA. Once hybridized, the tag is detected with a secondary antibody (e.g., anti-digoxigenin), which is linked to an enzyme that produces a visible chemical deposit on the cells. Horseradish peroxidase and alkaline phosphatase of the two most commonly used enzymes in this powerful approach, which is widely employed in cancer research. However, ISH is technically challenging and works best on mRNAs that are expressed in high abundance. For these reasons, the use of ISH in clinical laboratories is generally restricted to the assessment of kappa and lambda light-chain expression in cases of suspected myeloma, or to detect the presence of Epstein-Barr virus in cases of lymphoproliferative disease driven by this infection.
Fluorescence in Situ Hybridization (FISH)
Nucleic acid probes are used not only to detect mRNA, but to assess interphase chromosomes in tumor cells. The probes may consist of either DNA or RNA, and they vary in length from short oligonucleotides to multigenic chromosomal segments cloned into bacteria (so-called bacterial artificial chromosomes, or BACs). When directly labeled with a fluorophore, they can be detected using a microscope equipped for immunofluorescence. This approach is routinely used in clinical cytogenetic laboratories to assess gene copy number as well as gene translocations. For example, FISH is commonly used to evaluate breast carcinomas for amplification of the ERBB2 (HER2) locus and to screen for increased copies of NMYC in neuroblastoma and MET in non–small-cell lung carcinoma. Loss of tumor suppressors such as PTEN, CDKN2A, and TP53 can also be assessed by this approach.
There is a long and growing list of gene translocation events that are linked to cancer. Whether the result of intra- or interchromosomal exchanges, these translocations commonly involve genes encoding a kinase or a transcription factor. The resulting fusion genes are often the principal drivers of tumorigenesis and therefore serve as diagnostic markers and/or targets for specific therapies (e.g., kinase inhibitors). Fusion mRNAs from translocation events can be detected by highly sensitive methodologies based on polymerase chain reaction (PCR); however, these approaches can be frustrated by the fact that a particular target gene may be fused to any of more than a dozen different partner genes, requiring numerous primers to cover all possible fusion events. In contrast, FISH can detect a translocation-related break in a target gene irrespective of which partner gene has been fused to it. This is done by labeling two pools of probes with different fluorophores; for example, one pool may be labeled red and hybridizes to the 5′ end of the gene, while the other is labeled green and hybridizes to the 3′ end of the gene. If the gene is intact, the red and green signals are close together and merge into yellow, but if the gene has been split apart by a translocation event, the red and green signals become well separated (Figure 59-2 ). A further advantage of FISH is that only a few tumor cells are required to make a diagnosis, which is helpful in biopsies containing low tumor content.
Preparing Nucleic Acids from Cancer Specimens
Many of the assays that are used to molecularly subtype cancers require the extraction of either RNA or DNA from the tumor tissue. Two important issues are often overlooked in this regard. The first is that the nature of the tissue dictates the quality of the available nucleic acid. Whereas fresh-frozen tissue will yield RNA or DNA that is largely intact, the nucleic acids derived from FFPE tissue are highly degraded. Thus, assays meant for use on FFPE-derived RNA or DNA must take into account the short, fragmented nature of these derivatives.
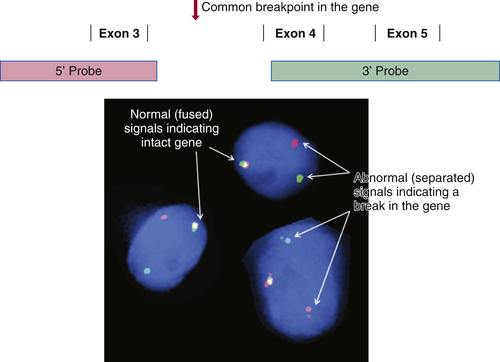
Figure 59-2 Fluorescence in situ hybridization (FISH) for identifying gene translocation events This approach is commonly used to look for evidence of gene translocations in interphase nuclei. Two probes are designed to hybridize to the 5′ and 3′ ends of a gene, flanking the region that is commonly “broken” during translocation. The probes are labeled with fluorescent tags of differing color, often creating a third color when they bind subjacently (e.g., green and red fusing into yellow). Wide separation of the probes indicates a break in the gene consistent with a translocation event. Note that this approach does not identify the partner gene in the translocation.
The second important issue is that tumor content and quality are highly variable from one specimen to the next, and even among different parts of the same specimen. For example, one paraffin block from a cancer resection specimen may consist of 90% tumor cells, while an adjacent block is dominated by stromal and inflammatory cells, with only 10% tumor cells being present. A third block from the same specimen may contain only necrotic tumor, as geographic necrosis is common at the center of large, high-grade malignancies. Thus, it is critical that all tumor material being considered for nucleic acid extraction first be evaluated by a qualified pathologist, who can assess whether the tumor sample is suitable for the proposed testing.
Tumor Enrichment by Macrodissection
The sensitivity of molecular diagnostic assays for mutations and other alterations in tumor DNA is dependent in part on the ratio of tumor DNA to normal cellular DNA present within a specimen. If 90% of the cells within a tissue sample are cancerous, mutation detection is straightforward, but if only 10% of the cells represent the tumor, DNA from the normal cells will effectively dilute out the mutation and make it harder to detect. For this reason, it has become standard practice to dissect out the tumor-rich areas of a specimen. This can be performed under a dissection microscope, but is more commonly done by simply scraping the tumor from unstained paraffin sections using a scalpel blade, or by taking a small core directly out of the paraffin block. Comparison with an H&E-stained section serves to guide the dissection.
Tumor Enrichment by Laser Capture Microdissection
Some tumor samples have too little tumor to allow macrodissection. In these cases, a microscope equipped with a laser can be used to isolate small clusters of tumor cells, or even single cells, from adjacent normal tissue elements. Although this is labor intensive and yields relatively little material for further testing, it can be used to salvage cases when no other tumor material from a patient is available.
Assays for Single Genes or Single Mutations
Over the past two decades, a long and growing list of genes that play a significant role in cancer has been identified. For mutations in those genes that are of particular prognostic importance or serve as important predictors of therapeutic response, a variety of assays have been developed for use in clinical laboratories. Most of these assays are focused on a single mutation or a set of mutations occurring in a single gene exon. Important factors in the design of such an assay include the amount of input DNA or RNA needed, the ease and rapidity with which the assay can be completed, and its sensitivity and specificity. Of course, assay cost is another important factor. There are many different platforms used to support these types of assays, and each represents a significant investment for the laboratory.
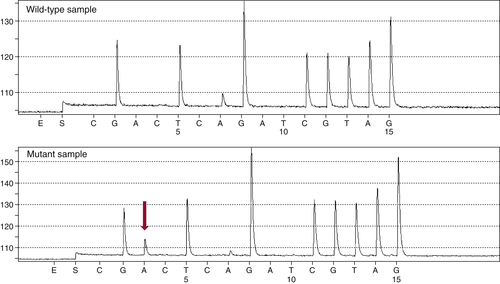
Figure 59-3 Pyrosequencing Two pyrograms are shown from tumor samples subjected to pyrosequencing for common mutations in the KRAS gene. The presence of an extra A peak in the lower panel is evidence for the presence of a mutation; the preceding G peak has a correspondingly lower signal, indicating a G→A substitution.
Sanger Sequencing
The single most common approach to screening mutations in cancer genes is Sanger sequencing. First described by Fred Sanger and colleagues in 1976, this method mixes nonextendable, fluorescently labeled dideoxy nucleotides together with standard nucleotides to generate a set of fragments of varying length as the original template DNA is copied. 11 When separated by capillary electrophoresis, the sequence of the DNA can be interpreted from the color of the last incorporated (dideoxy) nucleotide on each successive fragment. Highly reliable and reproducible, Sanger sequencing was used to generate the first complete sequence of the human genome. 12 Nevertheless, it has several significant drawbacks. First, it can only detect mutations that are present in at least 15% to 20% of the input DNA molecules. Second, it takes approximately 24 hours to perform and is relatively labor intensive. Third, it can only cover mutations across approximately 200 bp of FFPE DNA. This means that many genes require multiple, overlapping sequencing reactions in order to ensure that a mutation can be identified, adding to the overall cost. For this reason, many laboratories rely on other approaches for screening common cancer-related mutations.
Pyrosequencing
Developed as a rapid and sensitive approach to sequencing very short regions of DNA (up to approximately 30 bp), pyrosequencing is used by many laboratories to screen for common mutations in tumor oncogenes such as KRAS, EGFR, and BRAF. 13 This technology differs from Sanger sequencing in that it does not require the synthesis and electrophoretic separation of DNA fragments. Instead, the incorporation of nucleotides during DNA synthesis is measured through a secondary reaction: each pyrophosphate released during nucleotide incorporation results in the generation of a light signal by the enzyme luciferase. As dATP, dCTP, dGTP, and dTTP are sequentially added, their incorporation is detected by the luciferase reaction, allowing the DNA sequence to be interpreted (Figure 59-3 ). The principal advantages of pyrosequencing are speed (sequencing can be completed in 2 hours) and sensitivity (as low as 5% mutant allele). A modification of the method is commonly used to assess DNA methylation.
High-Resolution Melting Curve Analysis
First developed in the late 1990s by Wittwer and colleagues, high-resolution melting curve analysis is now the backbone of a variety of assays designed to quickly and cost-effectively identify the presence of a mutation. 14 After the DNA of interest is amplified by standard PCR, the melting analysis can be performed in approximately 20 minutes without the need to add reagents or switch to a different instrument. The analysis begins by melting apart the DNA strands of the final DNA product at 95° C and then allowing them to re-anneal as they cool. When a mixture of wild-type and mutant DNA is present in the sample, heteroduplexes are formed, and these can be detected by slowly ramping the temperature back up to 95° C in the presence of a fluorescent dye that binds preferentially to double-stranded DNA. As the temperature increases, the heteroduplexes unravel at a slightly lower temperature than the homoduplexes of wild-type/wild-type or mutant/mutant DNA (Figure 59-4 ). Thus, the presence of a mutation shifts the melting point of the overall population, and this shift is readily detected by a decrease in fluorescence as the DNA strands separate.
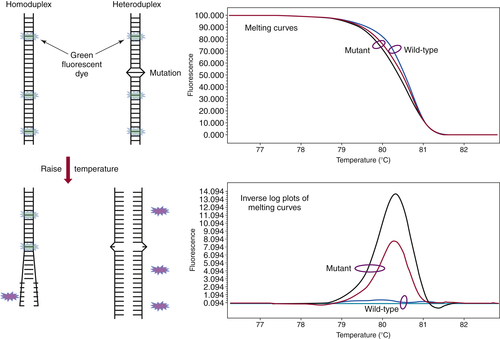
Figure 59-4 High-resolution melting curve analysis This method takes advantage of the fact that DNA heteroduplexes formed when a mutation is present have a lower melting temperature than do homoduplexes. The resulting shift in the melting curve can be detected by loss of fluorescent signal as a dye intercalated into the double-stranded DNA is released. Plotting the inverse log of the slope of the melting curve highlights the differences between samples that are wild-type and those containing a mutation. The approach can be used to detect point mutations, insertions, and deletions within amplicons of up to several hundred base pairs.
A number of variations of this assay type have been developed. Some will allow exact identification of a specific mutation, whereas others are optimized to detect intragenic deletions or insertions, the sequence of which requires confirmatory Sanger sequencing. In general, the sensitivity of these assays is approximately 10% mutant allele.
Allele-Specific PCR
A number of approaches have been developed that promote selective PCR amplification of a mutant allele over a wild-type allele. 15 Each depends on a primer that selectively binds to a site of mutation, the specificity being determined by the degree to which the mutant allele is favored over the wild type. Modifications to the primer, such as inclusion of peptide nucleic acids or locked nucleic acids, are generally necessary to achieve the selectivity that is optimal for clinical use. 16 The best assays have detection levels below 1%, with virtually no background contributed by false priming of the wild-type allele.
Multiplexed Approaches to Cancer Genotyping
The past decade has witnessed an explosion in the number of genes identified as playing a causal role in tumor development. This growing list of genes presents a challenge to clinical laboratories, which have traditionally used single-gene or single-exon approaches to genotyping. An alternative is to screen multiple genes/exons simultaneously using a multiplexed method, thereby saving on the labor and cost of performing multiple Sanger sequencing reactions. Two platforms have been developed to rapidly and cost-effectively screen for up to several hundred cancer gene mutations simultaneously. Both of these platforms—SnaPshot assays and mass spectroscopy–based assays—are based on so-called primer extension reactions. 17 In brief, each gene/exon of interest is amplified by standard PCR and then allowed to anneal to an oligonucleotide primer that sits immediately adjacent to a potential site of mutation. Dideoxy nucleotides are then added, and the primer is extended by a single base (essentially a single-base sequencing reaction), following which the product is interrogated to determine whether the added base represents wild-type DNA, a mutation, or both (Figure 59-5 ). The power of this approach is that the analysis can be performed in a multiplex manner, allowing many mutation sites to be screened at the same time.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


