Figure 35-1 Pancreatic epithelial neoplasia and the multistep model of exocrine pancreatic cancer. (From Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902-912, with permission.)
Table 35-1
Frequent Molecular Alterations in Pancreatic Adenocarcinoma
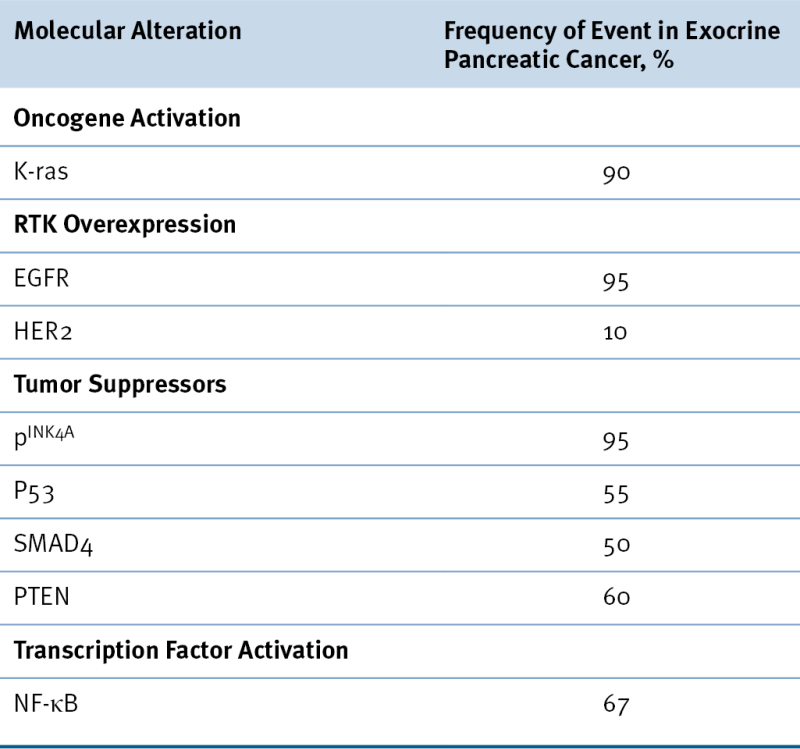
RTK, Receptor tyrosine kinase.
To overcome these effects, a second mutation, such as inactivation of tumor suppressor genes, p16, SMAD4, and/or p53 may be necessary for the development of invasive/metastatic pancreatic adenocarcinoma. Consistent with this hypothesis, more recent transgenic models have used genetically engineered mice with promoters that are developmentally expressed in progenitors of all pancreatic cell types. For example, genetically engineered mice expressing the mutant K-ras(G12D) allele mutation develop focal premalignant lesions consistent with human PanIN, 8 but a mouse with activation of a mutant K-ras allele (Kras(G12D)) and deletion of a conditional Ink4a/Arf tumor suppressor allele resulted in an earlier appearance of PanIN lesions, and these neoplasms progressed rapidly to highly invasive and metastatic cancers. 9 Similarly, the KPC mouse model was created by interbreeding mice with mutant K-ras(G12D) and Trp53(R172H) alleles with the PDX-1-Cre transgenic mice. These mice developed pancreatic adenocarcinoma rapidly and demonstrated a widely metastatic phenotype with a markedly shorter life span. 10 The evolution of these tumors bears striking resemblance to the human disease, possessing a proliferative stromal component and ductal lesions with a propensity to advance quickly. These findings in the mouse provide experimental support for the widely accepted model of human pancreatic adenocarcinoma in which activated K-ras serves to initiate PanIN lesions, and other tumor suppressors function to constrain the malignant conversion of these PanIN lesions into lethal ductal adenocarcinoma.
Figure 35-2 Major pathways and processes that are genetically altered in most pancreatic cancers. From Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-1806.
Recently, an inducible, genetically engineered mouse model of oncogenic K-ras revealed the integral role that K-ras plays in tumor maintenance and metabolism. 11 This mouse model supports the view that advanced pancreatic cancer remains dependent on K-ras expression. Furthermore, transcriptomic and metabolomic analyses demonstrated that mutant K-ras controls glycolysis by regulating glucose transporters as well as shunting glucose metabolism to anabolic pathways. These findings demonstrate how K-ras modifies metabolic pathways in pancreatic tumors to support the high energy requirements of tumor metabolism. These metabolic targets may also serve as novel targets for pancreatic cancer therapy.
Although K-ras mutation appears critical to the initiation of human pancreatic carcinogenesis and the initiation and maintenance of pancreatic cancer in genetically engineered mouse models, its role in the maintenance of established human pancreatic adenocarcinoma remains less clear. In addition, although K-ras mutation is widely detected in pancreatic adenocarcinoma, its expression can also be detected in nonmalignant conditions such as chronic pancreatitis. Disappointingly, novel therapies that target mutant K-ras have not been effective.
Epidermal Growth Factor Receptor
The role for epidermal growth factor receptor (EGFR) and its downstream signaling molecules in tumorigenesis is evidenced by their ability to transform normal cells to a neoplastic phenotype when expressed in mutated, unregulated forms or when expressed to an abnormally high level. Overexpression of EGFR and its downstream signaling molecules occurs frequently in a variety of human cancers, including pancreatic cancer. A prospective study indicated that EGFR was detectable in more than 95% of patients with advanced pancreatic cancer. 12 In most cases, EGFR is concomitantly expressed with its ligands, EGF or TGF-β. It has been hypothesized that the increased expression of ligand and receptor forms an autocrine loop that constantly stimulates cell proliferation. A study found that pancreatic cancer cell lines display heterogeneous sensitivity to the EGFR inhibitor gefitinib. Three of nine cell lines studied displayed significant sensitivity to pharmacologically relevant concentrations of gefitinib (1 μmol/L) as measured by two independent assays for G1-S cell cycle arrest. 13 Clinically, erlotinib, an oral small-molecular inhibitor of EGFR, was approved for the treatment of advanced pancreatic cancer based on a study by Moore and colleagues. 14 In this study, 569 patients were treated with either gemcitabine alone or gemcitabine with daily erlotinib. Median overall survival was significantly improved with the addition of erlotinib (6.24 months vs. 5.91 months, P = .038). Although the incremental survival benefit is small, it does suggest a role for the EGFR pathway in a subset of patients with pancreatic cancer.
Activation of Nuclear Transcription Factors
Nuclear Factor κB
Nuclear factor κB (NF-κB) is a family of pleiotropic transcription factors that regulate the expression of a spectrum of genes important in growth, oncogenesis, differentiation, and apoptosis. NF-κB proteins are normally sequestered in the cytoplasm in an inactive form through their association with the inhibitor IκBα, which masks the nuclear localization signal (NLS) of NF-κB, thereby preventing its nuclear translocation. NF-κB is activated through activation of the IκB kinase complex (IKK), which phosphorylates IκBα and, as a result of proteasomal degradation, releases NF-κB from the complex, exposing the NLS. Constitutive NF-κB activation has been detected in approximately 70% of pancreatic adenocarcinomas and 9 of 11 human pancreatic tumor cell lines, but not in normal pancreatic tissue. 15
Many upstream events can potentially affect NF-κB and other transcription factors. PTEN loss in conjunction with oncogenic K-ras activates NF-κB and its vast cytokine network. This results in a stromal response and inflammatory infiltration with tumorigenic potential. 16 Furthermore, proinflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin 1 induce rapid degradation of IκBα, resulting in nuclear translocation of NF-κB and sustained activation of NF-κB mediated through IκBβ. The EGFR-mediated signaling cascade and Ras signaling pathways may induce constitutive NF-κB activity as well.
Once activated, NF-κB mediates transcription of numerous genes encoding growth factors, cytokines, and apoptotic and cell cycle regulators. The expression of the apoptosis inhibitors that are regulated by NF-κB include c-IAP1, C-IAP2, Traf1, Traf2, A20, IEX-1L, and the Bcl-2 homologues Bfl-1/A1 and Bcl-x. NF-κB also activates the expression of genes that are important in invasion and metastasis, including matrix metalloproteinases, urokinase plasminogen activator, and ICAM-1. Like NF-κB, numerous other nuclear transcription factors such as AP1, 17 Sp proteins, 18,19 and Stat3 20 have been shown to be activated in exocrine pancreatic cancer. In some cases these proteins are being actively targeted for therapy. 21
Loss of Tumor Suppressors
INK 4 A and ARF Tumor Suppressors
Studies have demonstrated that the 9q21 locus encodes two important and overlapping tumor suppressor proteins: p16(INK4A) and p19(ARF). Loss of p16(INK4A) appears to be critical for the development of exocrine pancreatic cancer, occurring in up to 95% of cases. 22 Genetically, loss of the INK4A locus can occur through mutation, deletion, or promoter hypermethylation. Functionally, loss of p16(INK4A) allows CDK4/6 to phosphorylate RB, thereby facilitating entry into the S-phase of the cell cycle. The importance of p16(INK4A) in pancreatic carcinogenesis has been highlighted by studies of Bardeesy and co-workers, where p16(INK4A) mutations cooperate with mutant K-ras and p53 mutations in the development and progression of exocrine pancreatic carcinomas. 23
p53
The p53 tumor suppressor is mutated in at least 50% of patients with exocrine pancreatic cancer and appears to be most important during the later stages of tumor progression. 24 Loss of p53 may also contribute to the drug resistance and chromosomal instability that are characteristic of pancreatic cancer.
SMAD4
SMAD4 is a tumor suppressor gene. 25 Smad proteins belong to a family of proteins that are part of the TGF-β signaling pathway and negatively regulate the growth of epithelial cells. On binding of TGF-β, TGF-β receptor II activates TGF-β receptor I by phosphorylation. TGF-β receptor I in turn activates Smad2 and Smad3. The activated Smad2 and Smad3 form a hetero-oligomer with Smad4. This Smad complex translocates to the nucleus, where it interacts with DNA directly or indirectly through other DNA-binding proteins, regulating transcription of target genes. SMAD4, also known as DPC4 (homozygously deleted in pancreatic carcinoma locus 4), is frequently deleted or mutated in pancreatic carcinoma. Nearly 90% of pancreatic carcinoma cases show loss of heterozygosity for SMAD4, and 30% to 37% have a homozygous deletion of the SMAD4 region. In addition, there are intragenic inactivating mutations, including nonsense, missense, and frameshift mutations. In total, approximately 55% of pancreatic carcinomas have deletion or an inactivating mutation of SMAD4. Loss of SMAD4 occurs with a frequency of 10% or less in other malignancies, which suggests a specific role for SMAD4 in pancreatic carcinogenesis. 26 Inactivation of the SMAD4 gene correlates with loss of expression of its protein and can be monitored during progression of PanINs. In one study, of 188 PanIN lesions examined, Smad4 was not expressed in 31% of the high-grade lesions, whereas all low-grade PanIN lesions had detectable Smad4 protein. 27 This observation is consistent with the notion that K-ras is the initiation factor in pancreatic carcinogenesis followed by alterations of a variety of genes, including SMAD4, p16, p53, and others.
SMAD4 may also be a prognostic factor. Using immunohistochemistry, the SMAD4 protein status of 249 pancreatic adenocarcinomas from patients who underwent pancreaticoduodenectomy was examined. The SMAD4 gene status of 56 (22%) of 249 pancreatic carcinomas was also determined. It was found that patients with pancreatic adenocarcinomas with Smad4 protein expression had significantly longer survival (unadjusted median survival was 19.2 months as compared with 14.7 months in patients with pancreatic cancers lacking Smad4 protein expression; p = 0.03). This Smad4 survival benefit persisted after adjustment for other known prognostic factors including tumor size, margins, lymph node status, pathologic stage, blood loss, and use of adjuvant chemoradiotherapy. 28 These findings may be explained by the correlation between SMAD4 status and metastatic burden described earlier in this chapter.
PTEN
The tumor suppressor gene PTEN is known to play a major role in embryonic development, cell migration, and apoptosis. 29 PTEN acts as a lipid phosphatase that regulates major signal transduction pathways and effectively inhibits phosphatidylinositol-3-kinase (PI3K)-mediated signaling. PTEN mutation, which occurs frequently in many solid tumors, is associated with constitutive activation of the PI3K/Akt pathway, resulting in tumors that are generally resistant to apoptosis. In pancreatic cancer, PTEN is not mutated but functionally abrogated through loss of expression. It was found that more than 60% to 70% of pancreatic cancer cell lines and tumor tissues have decreased or loss of expression of PTEN. 16,30 The role of PTEN in pancreatic carcinogenesis was also studied using a pancreas-specific PTEN knockout mouse model. 31 Knockout mice display pancreatic ductal metaplasia as the predominant phenotype and occasional PanINs. These lesions are characterized by progressive replacement of the acinar pancreas with highly proliferative ductal structures that contain abundant mucins and express Pdx1 and Hes1, two markers of pancreatic progenitor cells. A fraction of these mice develop ductal malignancy. Further studies showed that ductal metaplasia resulted from the expansion of centroacinar cells rather than transdifferentiation of acinar cells into ductal cells. These results suggest that PTEN actively maintains the balance between different cell types in the adult pancreas and that dysregulation of the PTEN pathway in centroacinar cells may contribute to the initiation of pancreatic carcinoma in vivo.
Reactivation of Developmental Biology Pathways
Hedgehog, Notch, and Wnt Pathways
The relationship between developmental pathways for pancreatic organogenesis and pancreatic cancer has recently gained in appreciation. 32 The hedgehog (Hh) family of genes—sonic hedgehog (Shh), Indian hedgehog (Ihh), and desert hedgehog (Dhh)—encode signaling molecules that regulate multiple functions during organ development and in adult tissues. Hedgehog signaling plays an important role in determining the fate of mesoderm in the primitive gut tube, as well as in early pancreatic development and islet cell function. Multiple groups have reported that the hedgehog, Notch, and Wnt developmental cascades might be reactivated during the development of pancreatic cancer through the upregulated expression and/or activation of these complex signaling pathways. 33 The hedgehog signaling molecules, Shh, Ihh, Ptc, Smo, and Gli1, are frequently overexpressed in pancreatic cancer tissues and cell lines as well as in PanIN lesions. 34,35 Specific inhibition of Hh activity in pancreatic cancer cells using cyclopamine can reduce pancreatic cancer cell growth both in vitro and in vivo (Figure 35-3 ). The reduction of the proliferative activity of pancreatic cancer cells is mediated through G0/G1 cell cycle arrest in vitro or induction of apoptosis in vitro and in vivo. Clinical studies with hedgehog inhibitors in pancreatic cancer are currently in progress. Results from these studies will better define the clinical relevance of this target in pancreatic cancer.
Downstream Events
To elucidate additional genetic alterations that can interact with K-ras in pancreatic carcinogenesis, the Sleeping Beauty transposon system has been recently employed in these K-ras mutant mouse models. Mann and colleagues performed a mutation screen using this system and identified 543 potential candidate cancer genes, of which 75 had known mutations in pancreatic cancer. 35 From this, they were able to categorize these genes based on function and clinical outcomes, providing a rich source of information on these potential targets. In a similar experiment, Perez-Mancera and associates used the Sleeping Beauty transposon system and identified certain driver mutations that overlapped with those found by Mann and colleagues. These mutations act in concert with K-ras to promote progression of pancreatic cancer. Interestingly, introduction of this inducible system also demonstrated a number of candidate genes for the development of pancreatic adenocarcinoma that were unique to this set of experiments. 36 In particular, the X-linked gene deubiquitinase Usp9x was inactivated in more than 50% of the tumors. Loss of this gene promoted cellular transformation and correlated clinically with poor prognosis, making it a potential target for future therapeutics.
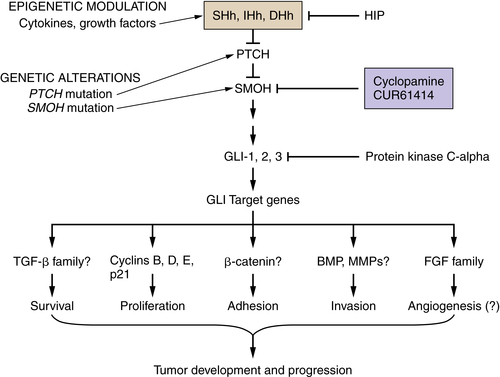
Figure 35-3 Hedgehog signaling, carcinogenesis, and potential therapeutic targets Upregulation of Hh ligands may be mediated by epigenetic events. Mutations in PTCH and SMOH result in activation of hedgehog signaling and are causative in basal cell carcinoma and medulloblastoma. Gli proteins are thought to mediate activation of Hh transcriptional targets potentially important in tumorigenesis, progression, and metastasis. DHh, Desert hedgehog; Gli, cubitus interruptus-like transcription factor involved in glioma formation; Hh, hedgehog; HIP, hedgehog interacting protein; IHh, Indian hedgehog; PTCH, patched; SHh, sonic hedgehog; SMOH, smoothened. From Xie K, Abbruzzese JL. Developmental biology informs cancer: the emerging role of the hedgehog signaling pathway in upper gastrointestinal cancers. Cancer Cell. 2003;4:245-247, with permission.
Desmoplastic Reaction (Tumor Stroma)
One of the morphologic hallmarks of pancreatic adenocarcinoma is its desmoplastic reaction, or tumor stroma. Desmoplastic tissue consists of fibroblasts (the main cellular component), infiltrating inflammatory and immune cells, endothelial cells, and extracellular matrix (ECM) proteins, such as fibronectin and collagen. 37 Pancreatic adenocarcinoma exhibits a threefold increase in interstitial fibrillar collagen (types I and III) compared with the normal pancreas. 38,39 The desmoplastic reaction is also associated with proliferation of fibroblastic cells, which in some cases outnumber tumor cells. Evidence suggests that these are mesenchymal cells, known as stellate cells, which have differentiated into an activated myofibroblastic phenotype. These activated myofibroblasts have been identified as the principal source of type I collagen in the desmoplastic stroma. 40 Pancreatic stellate cells are quiescent and can be identified by the presence of vitamin A–containing lipid droplets in the cytoplasm. In response to pancreatic injury or inflammation, pancreatic stellate cells are transformed from quiescent phenotypes into highly proliferative myofibroblast-like cells that produce ECM proteins. Activated pancreatic stellate cells are observed in abundance in pancreatic tumor tissue, suggesting that they are responsible for the deposition of matrix components and the desmoplastic reaction that surrounds the pancreatic tumor, although pancreatic tumor cells are also capable of producing ECM proteins. Cell culture experiments have demonstrated that pancreatic tumor cells stimulate the growth of pancreatic stellate cells and ECM formation. The growth-stimulating effects are probably mediated by platelet-derived growth factor, fibroblast growth factor 2, and TGF-β1 secreted by pancreatic cancer cells. On the other hand, pancreatic stellate cells can stimulate the growth of pancreatic cancer cells, as demonstrated by an in vivo study in which co-injection of pancreatic stellate cells and tumor cells subcutaneously produced larger and faster growing tumors than injection of pancreatic tumor cells alone. Pathologic examination of tumor tissues showed that an intense desmoplastic reaction in tumors developed after injection of pancreatic stellate cells and tumor cells. 41
TGF-β is one of the major growth factors stimulating the growth of pancreatic stellate cells. Evidence indicates that the predominant source of TGF-β may be infiltrating granulocytes, although pancreatic tumor cells are capable of producing TGF-β. 42 Immunohistochemical staining of pancreatic tumor tissue showed that isolated cells, mainly located at the invasive edge surrounding cancerous nests, prominently stained for TGF-β. Those cells contain a segmented nucleus and are negative for anti-macrophage (CD68) and positive for anti-granulocyte antibodies, indicating that they are granulocytes. 43
Hedgehog signaling has also been implicated in the dense stromal reaction associated with pancreatic cancer. A hedgehog paracrine loop between neoplastic cells and stromal cells promotes stromal desmoplasia. The genetically engineered KPC mouse model of pancreatic cancer that closely mimics the tumor-stroma relationship was evaluated to understand the interaction between the hedgehog pathway and the stromal barrier. 44 This model generated poorly vascularized and perfused tumors, similar to those seen in human pancreatic cancer. The researchers showed that drug delivery was impeded in this environment. Furthermore, response to gemcitabine was seen in only a small percentage of the tumors, which closely reflects what is seen clinically with this chemotherapeutic agent. The investigators subsequently demonstrated that a hedgehog pathway inhibitor, IPI-926, could disrupt the stromal barrier and enhance gemcitabine delivery to the tumor, thereby improving cytotoxicity and survival. Although the importance of the stromal barrier in chemoresistance needs to be better understood, stromal cells may be a potential target for future therapies.
Cytokine Production
Pancreatic cancer is known to secrete growth factors that stimulate cancer growth through paracrine or autocrine mechanisms. In addition, pancreatic cancer secretes many cytokines that affect cancer development through interaction with its microenvironment but also have an effect on overall host physiology. 45 Patients with pancreatic carcinoma often have elevated circulating levels of IL-6, IL-10, IL-8, and IL-1RA compared with the levels in healthy individuals. 46 Furthermore, elevation in one cytokine often correlated with elevation in others. For instance, high IL-10 levels were correlated with high IL-8 and high IL-6 levels. It was found that high IL-6 levels in patients with pancreatic carcinoma were correlated with worse survival and weight loss. 47 Further evidence suggested that IL-6 is involved in the development of cachexia, which is a clinical hallmark of pancreatic carcinoma. The pathobiology of cachexia is poorly understood; however, IL-6 and other cytokines may contribute to its development. 48 In one study, gene chip analysis of resected pancreatic cancer tissue including 5600 human genes revealed a significant difference between patients with and without cachexia in only four factors: IL-6, neuropeptide Y Y3 receptor, neurotensin, and islet amyloid polypeptide. IL-6 was significantly overexpressed in pancreatic specimens and elevated in the serum of cachectic patients. A coculture system revealed that pancreatic cancer cells can stimulate IL-6 production exclusively from peripheral blood mononuclear cells derived from cachectic patients, and this effect could be reduced by IL-6–neutralizing antibodies. These data indicate that IL-6 may represent a prominent cachexia-associated factor in pancreatic cancer.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


