Funding
This work was funded by Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Miguel Servet Type 2 contract (CPII18/00004 to J.J.), FEDER “Una manera de hacer Europa.” This work was supported by CIBER-Consorcio Centro de Investigación Biomédica en Red-CIBERDEM (leading group CB15/00071, D.M., and CB07/08/0016), Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación.
1
Introduction
Hyperglycemia is the consequence of uncontrolled glucose metabolism. Systemic elevations in glucose directly affect the physiology of a wide range of target tissues due to the intracellular accumulation of cytotoxic metabolites derived from an altered metabolism of glucose [ ]. Therefore, hyperglycemia is a contributing factor in the development of nonvascular and vascular pathologies.
Accumulating data from experimental studies have identified at least six main molecular mechanisms involved in glucose-mediated cell damage [ ]. Hyperglycemia contributes to intracellular metabolic abnormalities via the activation of a series of overlapping molecular mechanisms, including increased glycolysis, activation of polyol (sorbitol) pathway flux, increased glucose autoxidation, increased formation of advanced glycation end-products (AGE) and expression of the cognate receptors of AGE (RAGE), activation of protein kinase C (PKC) isoforms, and increased hexosamine biosynthetic pathway (HBP) flux ( Fig. 3.1 ) [ ].
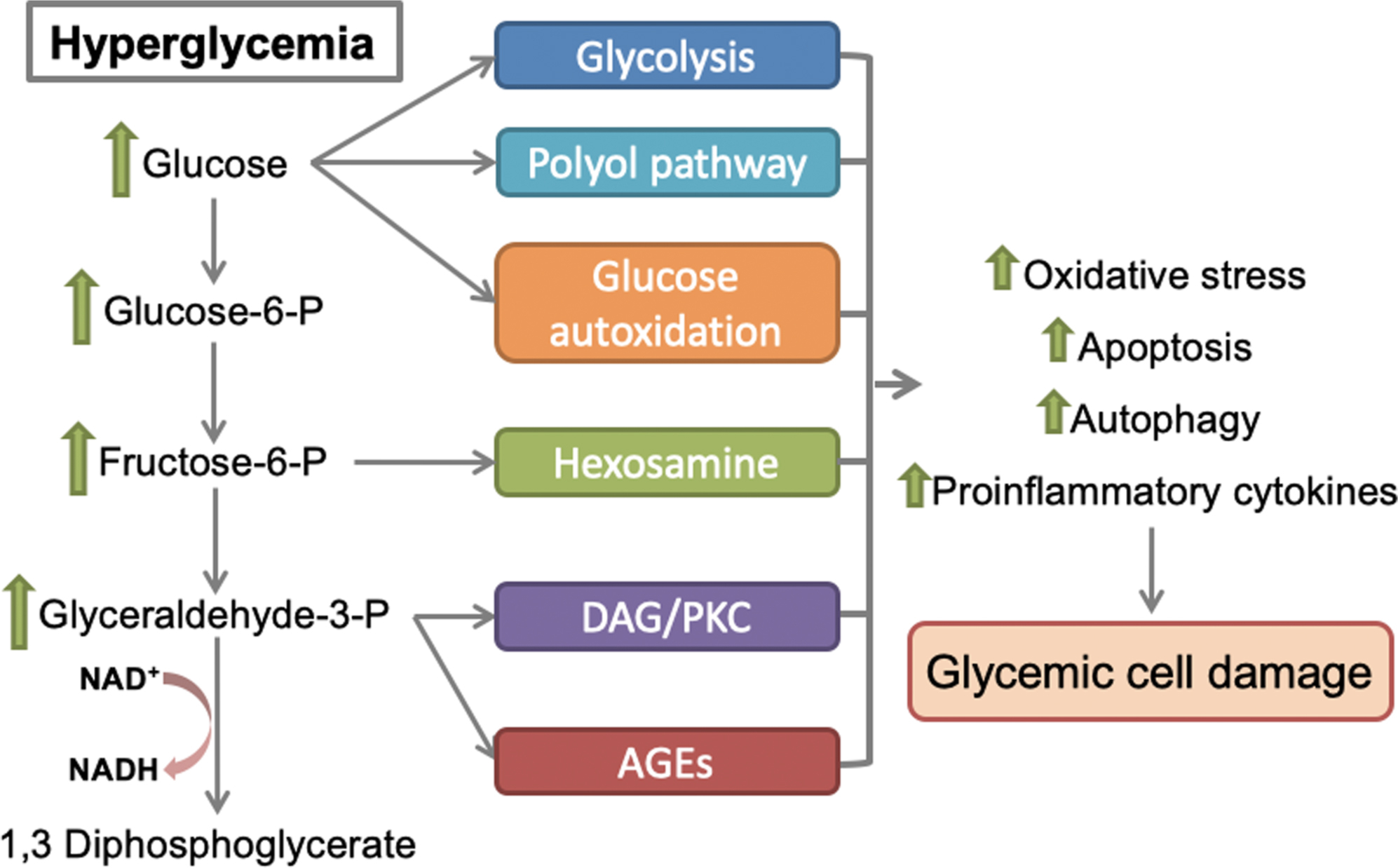
Oxidative stress is central in hyperglycemic states. The potential sources of hyperglycemia-induced oxidative stress are glucose auto-oxidation [ ], nonenzymatic glycation [ ], the interaction between glycated products and their receptors [ ], overproduction of reactive oxygen species (ROS) by mitochondria [ ], and the polyol pathway [ , ]. Under these conditions, the overproduction of ROS frequently overwhelms the capacity of the cellular antioxidant defense system. Increased oxidative stress affects the functional properties of different oxidized molecules and triggers the inappropriate activation of important regulatory molecules involved in a series of cellular responses that are involved in the pathogenesis of various diabetic complications [ ], including the activation of PKC [ , ], the nuclear factor kappa B (NF-κB) signaling [ ], and the JNK stress-associated kinases [ ].
Compelling data show that an increased formation of ROS/superoxide anion from mitochondria underlies the major cause of diabetes complications [ ]. Exposure to excessive glucose induces rapid mitochondrial dysfunction, which engages glucose metabolism in alternative pathways, primarily glycolysis, and HBP, which are frequently associated with the aberrant expression of AGEs/RAGEs to further promote oxidative stress and inflammation [ ].
We describe the main mechanisms involved in hyperglycemia-induced cellular damage. This chapter also deepens our understanding of the contribution of dysfunctional mitochondria to adverse cellular outcomes induced by glucose elevations.
2
Mechanisms of hyperglycemia-induced cell damage
The cellular damage induced by elevated levels of glucose may occur via several routes. Therefore, we focus on (i) increased nonoxidative glycolysis, (ii) increased activation of the polyol (sorbitol) pathway, (iii) glucose autoxidation, (iv) increased diacylglycerol (DAG)/PKC pathway, (v) increased formation of AGEs, and (vi) alterations in the HBP.
2.1
Increased glycolysis
Nonoxidative glycolysis, that is, glucose conversion to lactate, is markedly accelerated in hyperglycemic states [ , ]. Under these conditions, the glycolytic pathway is highly fluxed with high levels of glucose moieties [ ]. The increased flux of glucose to lactate is revealed by elevations in glucose-6-phosphate (G6P) synthesis, which increases flux into pathways that are not as representative in normoglycemic conditions, such as the hexosamine and pentose phosphate pathways. Notably, these elevations in lactate synthesis also lead to inadequate energy substrate utilization and impaired metabolic switching as observed in cultured skeletal muscle cells [ ]. This effect was specific to lactate because its addition produced the same effects as hyperglycemia on glucose metabolism and was not reproduced by any other organic acid in cultured myotubes [ ]. The increased lactate production may underlie the metabolic changes produced by hyperglycemia. In this regard, an increased glycolytic flux and preferential conversion of pyruvate into lactate may be explained by a decreased metabolism of pyruvate by the mitochondria.
2.2
Increased polyol (sorbitol) pathway flux
The polyol pathway is a two-step metabolic route in which excess glucose is reduced to sorbitol by aldose reductase (AR), which is a cytosolic, monomeric rate-limiting oxidoreductase. AR also catalyzes the reduction of numerous carbonyl compounds generated by ROS to inactive alcohols. The reduced form nicotinamide adenine dinucleotide phosphate (NADPH) acts as a cofactor in this reaction ( Fig. 3.2 ). The reduced product, sorbitol, may be further oxidized to fructose by sorbitol dehydrogenase, which is the second enzyme of this pathway and uses NAD + as a cofactor [ ].
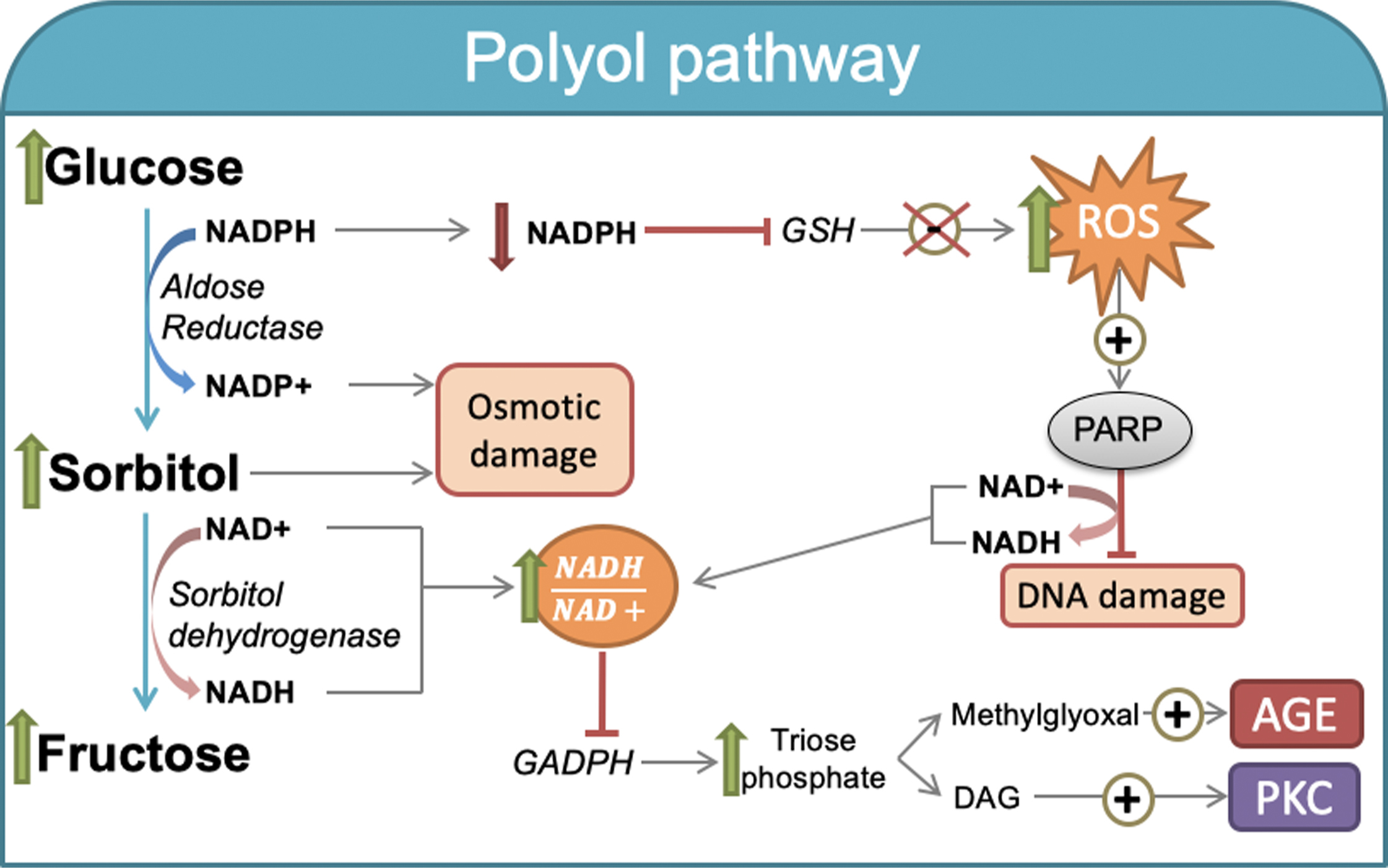
Because of the low affinity of AR for glucose under physiologically normal concentrations, cell glucose use is rather nominal. However, AR activity increases when intracellular concentrations increase, thereby accelerating the polyol pathway rate. Importantly, an increased polyol pathway also leads to concomitant reductions in intracellular NADPH and NAD + content [ ].
The rate of glucose flux through this pathway is species-, tissue-, and site-specific [ ]. Under hyperglycemic states, the sorbitol pathway is markedly accelerated, especially in the retina, kidney, peripheral nerves, and blood vessels, where insulin is no longer required for cellular glucose uptake; therefore, it may differentially contribute to the onset of tissue-specific diabetic outcomes.
The potential detrimental effects of the hyperglycemia-induced polyol pathway may be explained by different mechanisms. One mechanism is the osmotic stress caused by excess sorbitol accumulation. Supporting this notion, sorbitol may act as an intracellular osmolyte in certain cells, such as renal collecting duct cells, retinal pigment epithelial cells, and renal glomerular endothelium [ ]. The inability of intracellular sorbitol to diffuse across cell membranes may promote cellular damage due to alterations in their osmolarity. The polyol pathway was the first pathway linking hyperglycemia to microvascular complications [ , ]. Indeed, microvascular cells are particularly sensitive to the adverse consequences of increased sorbitol synthesis. In this regard, the toxicity of polyol accumulation in ocular lens cells is likely related to their active osmotic action. Increased intracellular accumulation of polyols leads to water influx, which promotes cell dysfunction. However, the content of sorbitol in diabetic vessels and nerves are so low, that sorbitol-mediated osmotic injury may not be relevant even under hyperglycemic conditions [ ].
The Na + /K + -ATPase activity rate is also commonly decreased upon high-glucose-mediated induction of polyol synthesis [ ]. Notably, the latter condition has further been linked to lower phosphatidylinositol synthesis, and it was shown recently to result from PKC activation.
Another potential mechanism is the oxidation of sorbitol in a reaction catalyzed by the enzyme sorbitol dehydrogenase. Consequently, the cytosolic NADH/NAD + ratio raises, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzyme activity decreases, and leads to a concomitant increase in the concentrations of triose phosphate [ ]. The latter has been accompanied by enhanced formation of a precursor of AGEs, methylglyoxal, and diacylglycerol (DAG), which further activates the PKC pathway. Increased ROS production under conditions of hyperglycemia, which positively induces poly(ADP-ribose) polymerase (PARP) activity (as discussed below). Because PARP is a NAD + -consuming enzyme, its induction may reduce tissue NAD + content and contribute to the increased intracellular NADH/NAD + ratio [ ]. Altogether, NAD + declines and the excess glucose being channeled through the polyol pathway [ ], further exacerbates oxidative stress under hyperglycemia.
The last mechanism potentially related to glucose-induced polyol metabolism is linked to NADPH depletion because AR consumes NADPH during the conversion of glucose into sorbitol ( Fig. 3.2 ). NADPH is necessary for regenerating the reduced glutathione (GSH) pools. GSH plays a critical role as an intracellular scavenger of ROS [ ]. Therefore, decreased NADPH may lead to decreased intracellular antioxidative activity, which increases intracellular oxidative stress and becomes the most likely mechanism by which hyperglycemia generates cell damage via the polyol pathway [ ].
Recent data support the role of the polyol pathway and the beneficial effects of AR inhibitors in the context of diabetic neuropathy, but clinical trials failed to demonstrate their effectiveness in humans [ ]. Greene and colleagues showed that treatment for 12 months with the AR inhibitor Zenarestat reversed the loss of myelinated nerve fibers in the sural nerve, which may stimulate regeneration but with adverse side effects [ ]. Therefore, the therapeutic modulation of AR activity is a challenging translational goal, and further research is deserved in this field.
2.3
Glucose autoxidation
Glucose may be autoxidized ex vivo in a chemical transition metal-catalyzed reaction that produces hydrogen peroxide (H 2 O 2 ), reactive intermediates, such as hydroxyl (OH − ), superoxide radicals (O 2 − ), and reactive carbonyl species (RCS), including ketoaldehydes [ ]. Glucose autoxidation occurs under conditions of excess glucose, and may be responsible for increased oxygen radicals in chronic hyperglycemia [ ]. The rate of RCS formation, including glyoxal, methylglyoxal, and 3-deoxyglucosone, is increased by several potential mechanisms, such as glucose autoxidation and glycoxidation of glucose-protein adducts [ ]. Although cellular carbonyl detoxification mechanisms mostly metabolize these species [ ], the enhanced formation of glyoxal, which is the major product of glucose autoxidation, may also contribute to extracellular protein damage [ , ].
Glucose autoxidation may also contribute to chemical modifications of extracellular proteins and promotes functional damage in vivo [ ]. Actually, glucose modifies lysine residues, but RCS reacts preferentially with arginine and lysine residues [ ]. The reactivity with arginine residues is of physiological importance because glyoxal, and methylglyoxal synthesis is increased under hyperglycemic conditions [ ] and may play an important role in the pathogenesis of excess glucose due to the elevated abundance of arginine residues within the active site of many proteins. In this regard, renal extracellular matrix proteins with a long lifespan may be particularly prone to glyoxal-derived modifications in critical arginine residues within the assembly interface of certain collagen IV domains in diabetic rats [ ].
2.4
Increased diacylglycerol/activation of the protein kinase C pathway
Hyperglycemia upregulates some isoforms of the mammalian PKC family (i.e., α, β, δ, and ε) with serine-threonine kinase activities [ , ]. Diacylglycerol (DAG) plays a key role in PKC isoform activation [ ]. The inhibition of GAPDH activity by hyperglycemia-induced oxidative stress promotes DAG synthesis, which leads to the activation of PKC isoforms [ ]. PKC activates the NADPH oxidase complex that contains multiple cytosolic components, for example, p47phox, p67phox, and p40phox. Upon activation, the NADPH oxidase complex translocates to the plasma membrane where it associates with NADPH oxidase (NOX)2 or cytochrome b -245 heavy chain to produce ROS [ ]. These signaling pathways link hyperglycemia with PKC-mediated ROS and proinflammatory cytokine production ( Fig. 3.3 ). The accumulation of ROS increases oxidative stress, promotes apoptosis, and exacerbates autophagy in cells exposed to hyperglycemic-mimicking conditions [ , ].
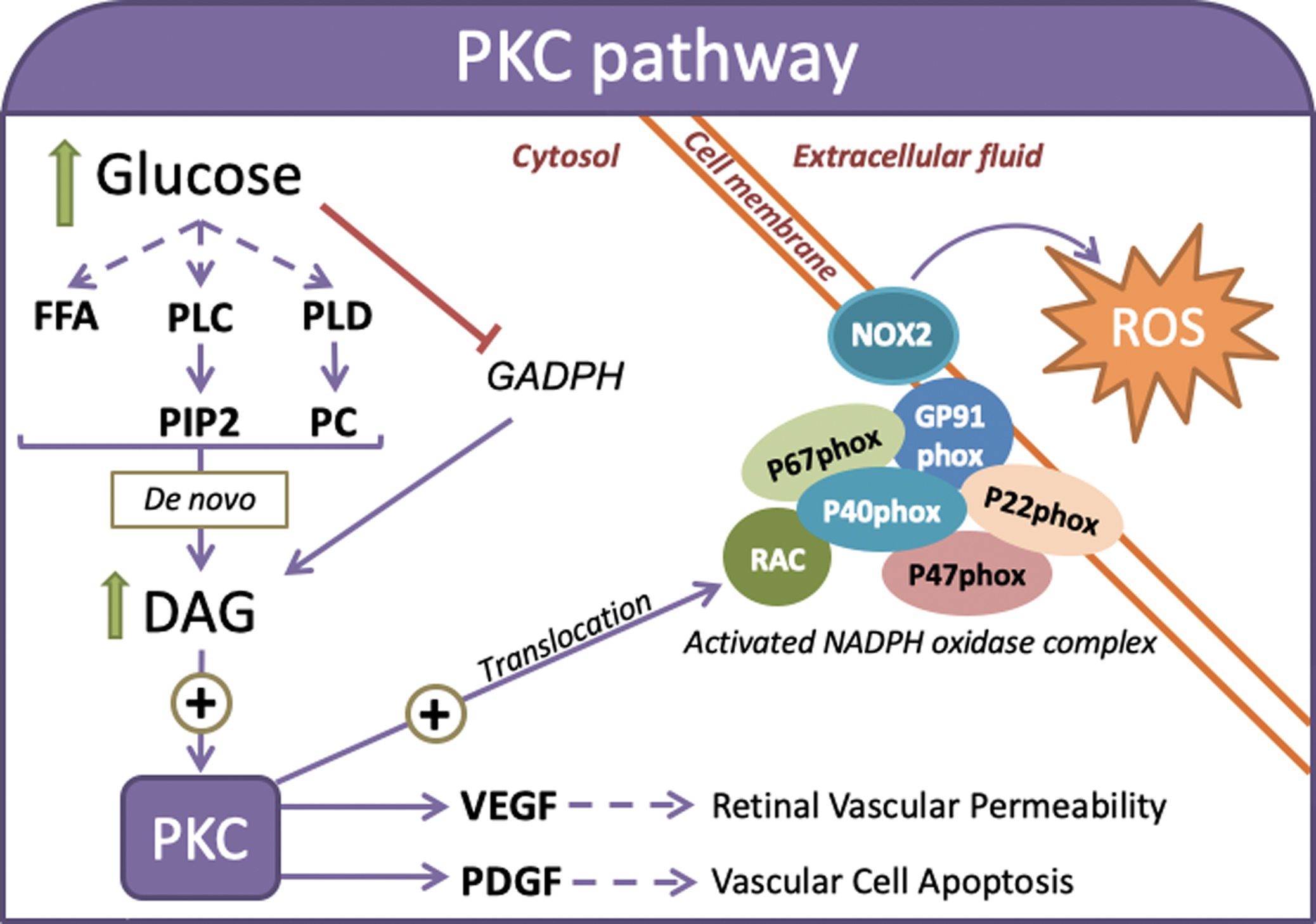
Retinal ganglion cells suffer oxidative stress-induced autophagy in response to multiple noxious stimuli. For example, PKC has been linked to microvascular damage in the diabetic retina because it promotes the synthesis and activation of mitogen-activated protein kinase (MAPK) and concomitantly increases the expression of Src homology-2 domain-containing phosphatase-1 (SHP-1). These PKC-mediated changes induce the dephosphorylation of platelet-derived growth factor (PDGF) receptor-β and alters its downstream signaling, which enhances vascular cell apoptosis [ ]. PKC activation may also induce the expression of vascular endothelial growth factor (VEGF) to regulate hyperglycemia-induced retinal vascular permeability [ ].
PKC-based therapeutic strategies to prevent diabetic retinopathy were evaluated in experimental studies and clinical trials. Notably, oral administration of ruboxistaurin and PKC-412, which downregulate PKC-β, successfully prevented visual loss and diabetic microangiopathy [ , ].
Glucose-induced PKCβ activation and ROS production are accompanied by an inhibition of nitric oxide (NO) production in smooth muscle cells [ ]. Consistent with these findings, hyperglycemia activates PKCβ, ICAM-1, and VCAM-1 and downregulates endothelial NO synthase (eNOS) activity, which promote macrophage adhesion to endothelial cells (EC) [ , ]. Hyperglycemia directly increases vascular permeability factor (VPF) expression, which induces angiogenesis and increases endothelial permeability in human smooth muscle cells in a PKC-dependent mechanism [ ]. NFκB activation also occurs in cultured ECs in response to the induction of PKC by glucose [ ]. Importantly, antidiabetic agents, such as metformin and liraglutide, inhibit hyperglycemia-induced ROS formation primarily by preventing intracellular DAG accumulation, which leads to a downregulation of the PKCβ-dependent NADPH oxidase complex in ECs [ ]. Overall, these findings strongly indicate that PKCβ plays a role in endothelial dysfunction by enhancing inflammatory responses to hyperglycemic states in vivo, which lead to accelerated diabetes-associated atherosclerosis.
2.5
Increased intracellular formation of advanced glycation end-products
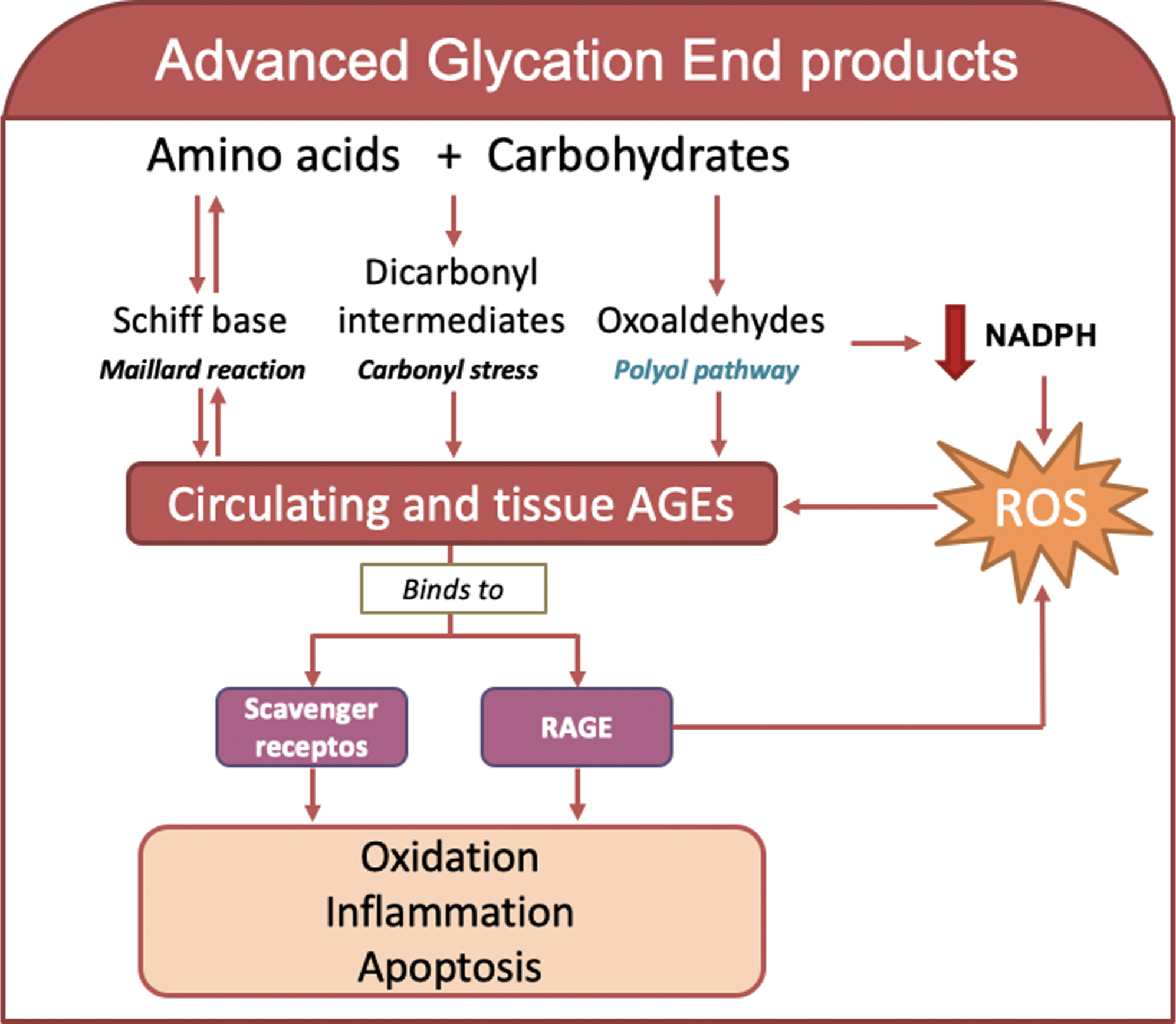
The accumulation of AGEs is an irreversible change under hyperglycemic conditions [ ]. Increased synthesis of AGEs and their signaling via RAGE lead to vascular dysfunction and end-organ damage [ , ] ( Fig. 3.4 ). AGE accumulation is determined by AGE synthesis, which is increased during hyperglycemia, and clearance (e.g., renal), frequently impaired in diabetic subjects. Cell dysfunction is frequently related to AGE accumulation and underlies the pathophysiology of chronic diabetic complications [ ].
2.5.1
Synthesis of advanced glycation end-products
AGEs are a large group of heterogeneous and highly reactive compounds that are synthesized via different pathways [ , ]. The formation of AGEs may be initiated by the spontaneous, nonenzymatic reaction of glucose with free amino groups in amino acids to form reversible intermediates of a Schiff base as an early glycation product and an Amadori product (e.g., HbA1c). These reactions are also known as the classic Maillard reaction. Alternatively, AGEs are also formed via carbonyl stress, when the oxidation of sugars and/or lipids boost the generation of dicarbonyl intermediates and use the highly reactive carbonyl groups to react with amino acids during AGE formation [ , ]. AGEs are also synthesized from oxoaldehydes (3-deoxyglucosone, methylglyoxal, and α-dicarbonyl) derived from the nonoxidative rearrangement of Amadori adducts from fructose-3-phosphate in the polyol pathway, as discussed earlier as one of the mechanisms of hyperglycemia-induced damage in diabetes. The accumulation of methylglyoxal and 3-deoxyglucosone, which are produced in the early stages of glycation processes, are recognized sensors of AGE formation. The reaction in the polyol pathway mediated by AR may further lead to concomitant NADPH and glutathione depletion, which contribute to increased oxidative stress and AGE production [ ]. Alternatively, the oxidation of other substrates, such as lipids, connective tissue in the extracellular matrix, nucleic acids, and monosaccharides, for example, glucose, fructose, glyceraldehyde, and glucose-derived carbonyl precursors, may also be used for irreversible AGE formation [ ]. In contrast, some immune cells, such as neutrophils, monocytes, and macrophages, produce myeloperoxidase and NADPH oxidase enzymes upon inflammation, which contribute to the oxidation of amino acids and AGE formation [ , ]. AGE-bound RAGE in combination with ROS generation may enhance AGE production via the NADPH oxidase pathway [ , ]. Notably, the nuclear protein high-mobility group box 1 (HMGB1) may activate RAGE [ ], which further contributes to tissue inflammation [ , ]. The accumulation of AGEs and their functionally compromised adducts on nucleic acids, lipids, and proteins was recently proposed as a new marker of many age-related morbidities, including cardiometabolic diseases, such as diabetes [ ].
2.5.2
Receptors of advanced glycation end-products
AGE-modified molecules specifically or nonspecifically interact with several receptor systems, for example, scavengers, receptors, and interactions. The binding of AGEs to their receptors produces favorable or negative biological effects. Collectively, the clearance of AGEs by its receptors may ameliorate cellular AGE-induced oxidative stress and toxicity or activate a stress response that leads to inflammation and concomitant cellular dysfunction [ ].
The binding of AGEs to RAGE primarily mediates cellular AGE toxicity. Other AGE receptors (e.g., AGE-R1, R2, and R3) are also involved in the clearance of AGEs. AGE-R1 and AGE-R3 exhibit high-affinity binding to AGE ligands, and AGE-R2 shows signal transduction properties upon AGE binding. The downregulation of AGE-R1 expression has been linked to tissue elevations of AGE and impaired renal function in experimental mice [ ], and its upregulation increased the clearance and degradation of AGE and protected against AGE- or RAGE-induced cellular toxicity [ ]. Mice with AGE-R3 deficiency are more prone to AGE-induced glomerular damage [ ]. Conversely, AGE-R2 may elicit an inflammatory response [ ]. Other cell-associated receptors, including RAGE, scavenger receptor class A type II, MSR, and class B type I CD36, also interact with AGE derivatives and may promote inflammation [ ].
Lysozymes are another player in AGE clearance [ ]. This host-defense, antimicrobial protein exhibits a high affinity for AGEs, specifically for reactive Nε−(carboxymethyl)lysine and methylglyoxal precursors [ ]. Lysozymes remove in vivo-derived toxic AGEs from serum samples of diabetic and uremic subjects [ ]. Lysozymes also reduced systemic AGEs and enhanced their renal clearance in vivo, suppressed AGE-induced TNFα production by macrophage-like cells, and improved diabetic renal damage [ ].
2.5.3
Molecular effects of advanced glycation end-products
Advanced glycation is particularly dangerous because AGEs modify the structure and function of proteins and macromolecules to negatively affect their physicochemical characteristics and impair their cellular functions. Some AGEs also form stable cross-linked structures with other molecules [ ], especially proteins with a longer lifespan, including constituents of the connective tissue extracellular matrix and vascular basement membranes [ , ], which are highly prone to AGE-mediated crosslinks. However, accumulating evidence also suggests that these structures are formed with molecules with a shorter lifespan, such as systemic proteins and lipids, which are significantly elevated in diabetic subjects and patients with kidney disease [ , ].
A representative example of the clinical significance of AGE modifications is the advanced glycation of the lipid component of LDL [ ]. The amino groups in LDL phospholipids, for example, phosphatidylethanolamine and phosphatidylserine, provide target sites for glucose to form AGEs. Therefore, AGE-modified LDL is directly proportional to glucose levels [ ]. AGE-modified LDL is more prone to oxidation in vitro [ ]. During advanced glycation, fatty acids may be oxidized even in the absence of exogenous free radical-generating systems [ ]. The reaction of glucose with amino groups in phospholipids also occurs in vivo, which contributes to the advanced glycation of fatty acids and oxidative stress. The main protein component of LDL, apolipoprotein (Apo)B, and the amino acids lysine and arginine are also AGE targets. The circulating levels of AGE-modified ApoB are significantly elevated in diabetic subjects [ , ], due at least in part to an impaired clearance of glycated LDL. The increased production and reduced clearance of AGE-modified LDL likely promote its retention in the vessel walls via a process that is at least partially facilitated by an interaction with RAGE. The increase in AGE-modified LDL has been linked to accelerated vascular damage in diabetes [ ].
2.6
Increased flux via the hexosamine biosynthesis pathway
Under normoglycemia, only a small fraction of glucose (1%–5%) is metabolized via the HBP [ ] ( Fig. 3.5 ). However, elevations in intracellular glucose due to hyperglycemia and insulin resistance-induced excess fatty acid oxidation may drive excess glucose to produce glucosamine-6-phosphate [ ]. This step is catalyzed by glucosamine-fructose-6-phosphate amidotransferase (GFAT), which is the rate-limiting enzyme of the HBP [ ]. Glucosamine-6-phosphate is transformed via a sequence of reactions to obtain the major end-product of this pathway, uridine diphosphate N-acetyl glucosamine (UDP-GlcNAc) [ , ]. UDP-GlcNAc is an allosteric inhibitor of GFAT and the sugar nucleotide donor for O-GlcNAcylation [ ]. This reversible post-transcriptional modification is mediated by O-linked, N-acetyl glucosamine transferase (OGT) [ , ], and O-linked N-acetyl glucosaminidase (OGA) is responsible for the removal of O-GlcNAc [ ] ( Fig. 3.4 ). Interestingly, the O-GlcNAcylation process is carried on serine/threonine residues of the protein, which are well-known phosphorylation sites [ ].

Several studies showed a protective role of HBP. For example, HBP limited the entrance of calcium into the cytosol, promoted cell survival, and attenuated oxidative stress in cardiomyocytes [ ] and pancreatic beta-cells in cases of ischemia/reperfusion or trauma hemorrhage [ ]. HBP reduced inflammatory processes in vascular smooth muscle cells (VSMCs) [ ]. However, chronic activation is often accompanied by deleterious effects, which occurs with many of the stress-response pathways.
Different mechanisms may exert the hyperglycemic HBP-induced toxic effects. The first mechanism is a change in the glycosylation pattern of proteins. Increasing the amount of O-GlcNAc increases substrate availability, and proteins become highly glycated [ ]. For example, the elevated levels of O-GlcNAc in EC in diabetic humans and murine models are associated with endothelial reactivity dysfunctionality [ , ]. Increased O-GlcNAcylation in the extracellular matrix of VSMCs from diabetic mouse arteries is related to the overexpression of Runt-related transcription factor 2 (Runx2), which promotes calcification [ , ]. OGT and OGA are highly expressed in rat retinal neurons, and glucosamine-induced HBP induces apoptosis, which may occur via suppression of the neuroprotective effects of the insulin/Akt signaling pathway [ ]. Apoptosis in retinal pericytes (PC) occurs after chronic exposure to high glucose levels, and the O-GlcNAcylation modification of p53 may be the mechanism [ ]. At the metabolic level, the O-GlcNAcylation of FAT/CD36, which is responsible for 50%–80% of fatty acid uptake in the heart, may be responsible for the increased ATP production seen in diabetic hearts [ ]. Elevated levels of O-GlcNAcylation in components of the electron transport chain are also involved in the altered energetic substrate utilization by mitochondria in cardiomyocytes [ ].
Another mechanism is the competition of O-GlcNAcylation with phosphorylation sites. Phosphorylation and glycosylation are competing processes that strongly influence the action of endothelial nitric oxide synthase (eNOS). Indeed, early studies in bovine and human aortic ECs showed impaired eNOS activity after treatment with high glucose or hexosamine [ , ]. The increase in HBP reduces eNOS phosphorylation at Ser1177 (a known activation site for eNOS), which causes an abnormal aorta relaxation response [ , ]. eNOS is also a key regulator of renal function [ ]. It is activated via phosphorylation by protein kinase B (Akt), and Akt is activated by the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K). When a chronic increase in GlcNAc is present, a decrease in Akt/eNOS phosphorylation reduces the availability of NO in renal proximal tubular epithelial cells [ ], which promotes apoptosis and disturbs glomerular and tubular hemodynamics [ ].
Another case is the increased O-GlcNAcylation of thioredoxin interaction protein (TxNIP) in human and INS-1832/13 rat beta-cells, which enhances inflammasome activation [ ]. Cardiac-type sarcoplasmic reticulum Ca 2+ ATPase (SERCA2) is an enzyme that allows Ca 2+ entry in cardiomyocytes, and it is regulated by phospholamban (PLB) [ ]. SERCA2 activity is inhibited when PLB is not phosphorylated. Elevated O-GlcNAc levels competed with phosphorylation sites in PLB in isolated cardiomyocytes and in a mouse model of T2D [ ].
There are also modifications in key enzymes of the HBP. Overexpression of key enzymes from the HBP and their subcellular localization are altered in cases of hyperglycemia. For example, VSMCs were the first cell type in which the increase in Sp1 transcription factor O-GlcNAcylation, which was mediated by the overexpression of GFAT, was described and related to the glycemia-induced activation of plasminogen activator inhibitor 1 (PAI1), which is an inhibitor of fibrinolysis [ , ]. This effect was also described in ECs with the overexpression of TGF-beta [ ] and in fibroblasts, which may contribute to the overexpression of collagen [ ]. However, the chronic overexpression of GFAT in isolated rat pancreatic islets or treatment with hexosamine led to a deterioration of pancreatic beta-cell function via increased ROS production and a reduction in pancreatic and duodenal homeobox factor 1 (PDX-1) [ ]. The mislocalization of OGT and OGA in diabetic cardiomyocytes suggests a change in the O-GlcNAcylation pattern of susceptible proteins [ ].
3
Convergent element linking hyperglycemia-induced damage: Mitochondrial dysfunction?
Different stimuli influence mitochondrial biology, including mitochondrial respiration efficiency and mitophagy. Several studies revealed a link between alterations in mitochondrial dynamics and diabetes [ , ].
Convergent lines of evidence further suggest that hyperglycemia-mediated mechanisms are intricately interconnected and may be related to mitochondrial dysfunction [ ]. Several agents, for example, inhibitors of AGE formation, mitochondrial complex II, and uncouplers of oxidative phosphorylation, favorably influence cellular stress by reducing the effect of hyperglycemia on ROS production [ , ]. Hyperglycemia-mediated increased mitochondrial production of ROS/superoxide anion is functionally associated with the shunting of glucose into the HBP, which may induce long-term activation of proinflammatory signaling processes. Exposure of cultured cells to hyperglycemic conditions negatively influenced mitochondrial bioenergetics by enhancing the HBP-mediated inhibition of protein subunits of mitochondrial respiratory complexes I, III, and IV, which hampered cellular bioenergetics and ATP production [ ]. Different studies recently suggested a link between hyperglycemia-induced cell damage, mitochondrial dysfunction, NAD + decline, and impaired action of NAD + -consuming enzymes [ ]. NAD + reduction may result from several mechanisms in hyperglycemic conditions. First, the increased glucose flux into glycolysis and the Krebs cycle accelerates NAD + conversion into NADH [ ]. Massive NADH production via the polyol pathway (oxidation of sorbitol to fructose) may contribute to concomitant declines in cellular NAD + and reduction of the NAD + /NADH ratio [ ]. Noteworthy, abnormal reductions of the NAD + /NADH ratio have been defined as a state of pseudohypoxia [ ]. NADH elevations can raise glycerol-3-phosphate (a precursor for DAG), leading to enhanced superoxide production and oxidative stress. Chronic oxidative stress produces accumulating DNA damage, which leads to increased PARP activation [ , ] and an upregulation of CD38 expression [ ]. Because these enzymes are NAD + -consuming enzymes, their increased activity may also reduce NAD + pools and induce mitochondrial dysfunction [ ]. NAD + depletion may negatively influence another important class of NAD + -consuming enzymes, the NAD + -dependent histone deacetylases (also known as sirtuins) that catalyze the deacetylation of different substrates [ , ]. Upon deacetylation, some regulatory proteins that govern different cellular signaling pathways that are important in cell homeostasis may be activated. Accumulating evidence suggests that sirtuin 1 (SIRT1) is critical in alleviating mitochondrial dysfunction and oxidative stress [ , ]. Conversely, reduced SIRT1 expression or activity is a frequent feature in experimental conditions of hyperglycemia [ , ]. Other evidence also suggests a favorable role of SIRT1 in attenuating mitochondrial damage produced by hyperglycemia-mediated oxidative stress [ , , ]. Overall, these data suggest that mitochondrial dysfunction could be the common element underlying distorted cell homeostasis.
4
Concluding remarks
With the exception of muscle, liver, and myocardium, where insulin is needed for glucose metabolism, altered cell homeostasis in most tissues is directly related to insulin-independent mechanisms. Chronic exposure to high levels of glucose in these tissues directly leads to excess glucose supply and are classic sites of diabetic complications, such as the retina, kidney, neurons, and vascular EC [ ]. Therefore, persistent hyperglycemia is widely recognized as the causal factor in a wide range of reversible and irreversible effects that lead to progressive cumulative cell dysfunction. Chronic hyperglycemia has been functionally linked to the enhancement of several aberrant cellular mechanisms. First, the formation of AGEs, which via RAGE, contributes to oxidative stress and inflammation. Second, the selective enzymes of the HBP, which promotes the post-translational modification of several key cellular regulatory enzymes and membrane proteins, and finally, other underlying mechanisms of hyperglycemia-mediated effects, which include altered expression and signaling by PKC isoforms and increased flux through the aldose reductase pathway. Notably, accumulating evidence in the last decade revealed the contribution of a novel hyperglycemia-driven, ROS-increasing mechanism concomitant with dysregulated mitochondrial activity. Of particular translational interest are the favorable influence provided by SIRT1-increasing strategies in the amelioration of mitochondrial homeostasis and oxidative stress in hyperglycemic states [ , , ], which open new venues in the treatment of hyperglycemia-mediated complications of diabetes mellitus.
References
Related posts:
 Overview on chronic complications of diabetes mellitus
Overview on chronic complications of diabetes mellitus
 Epidemiology of chronic complications of diabetes: A global perspective
Epidemiology of chronic complications of diabetes: A global perspective
 Pathogenesis of diabetic atherosclerotic cardiovascular disease
DEFINITIONS
Pathogenesis of diabetic atherosclerotic cardiovascular disease
DEFINITIONS
 Metastatic Paraganglioma: Role for Systemic Chemotherapy
Metastatic Paraganglioma: Role for Systemic Chemotherapy
 Bilateral Macronodular Adrenal Hyperplasia in the Setting of Multiple Endocrine Neoplasia Type 1
Bilateral Macronodular Adrenal Hyperplasia in the Setting of Multiple Endocrine Neoplasia Type 1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


