(1)
Department of Patholog, University of Massachusetts Medical School, Baystate, Springfield, MA, USA
(2)
Department of Pathology and Laboratory Medicine, University of North Carolina School of Medicine, Chapel Hill, NC, USA
(3)
Department of Pathology and Laboratory Medicine, Hartford Hospital, Hartford, CT, USA
Keywords
Non-Hodgkin lymphomaHematologic malignanciesB-cell and T-cell developmentMolecular mechanismsMantle cell lymphomaFollicular lymphomaAnaplastic large cell lymphomaAdult T-cell leukemia/lymphomaMolecular profiling31.1 Introduction
Hematologic malignancies are a heterogeneous group of disorders that include neoplasms derived from cells of myeloid and lymphoid lineages. These malignancies can be categorized in many ways, but the most commonly used classification today is the World Health Organization’s Classification of Tumours of Haematopoietic and Lymphoid Tissues (WHO classification ) [1]. The WHO classification is largely based on the Revised European American (REAL) classification , which divided this diverse group of disorders into what were felt to be distinct or real clinicopathologic entities on the basis of combined morphologic, immunophenotypic, clinical, and genetic features [2]. The lymphoid neoplasms include immature B-cell and T-cell neoplasms (precursor lymphoblastic leukemia/lymphoma), mature B-cell and T-cell neoplasms (including the group of disorders collectively known as non-Hodgkin lymphomas (NHL) and plasma cell myeloma) and Hodgkin lymphoma. A review of Surveillance, Epidemiology, and End Results (SEER) data stated that in 2005 the United States would have approximately 93,420 incident cases of lymphoid neoplasms with approximately 38,000 deaths. Over the 10-year course of SEER data that were reviewed, the B-cell lymphomas showed a modest decrease in incidence (approximately 1 % per year), while T-cell neoplasms increased and Hodgkin lymphoma incidence remained stable [3]. Because lymphomas comprise a significant number of new cancer diagnoses, it is important to understand the pathogenesis of these disorders and how this information can be used for diagnosis, prognosis, and guidance of therapy.
The diagnosis of hematologic malignancies often requires a multi-parameter approach, correlating morphologic evaluation of traditional hematoxylin and eosin-stained tissue sections or Romanowsky-stained smears along with a variety of ancillary studies, including cytochemical and histochemical stains, immunohistochemical studies, flow cytometric analysis, cytogenetic analysis, and molecular genetic techniques. Of these, immunohistochemistry is favored among many pathologists because of the ability to directly correlate with the tissue morphology and convenience in working with readily available paraffin-embedded tissue (PET) (Fig. 31.1). Other methods may be more sensitive and quantitative than immunohistochemistry, but each method has advantages and disadvantages. For instance, flow cytometric analysis can detect small populations of aberrant B-cells and T-cells in a background of normal lymphocytes, a feat that is often not possible with routine immunohistochemical analysis. However, this technique requires viable cells in suspension for analysis and allows only limited correlation with morphologic findings. Molecular studies by polymerase chain reaction (PCR) or Southern blotting are very sensitive in fresh or frozen tissue, but sensitivity may be decreased in some assays using the more commonly available PET. Any of these ancillary studies can produce false positive and false negative results, and the interpretation of results must be made in the context of the traditional morphologic findings and clinical findings.
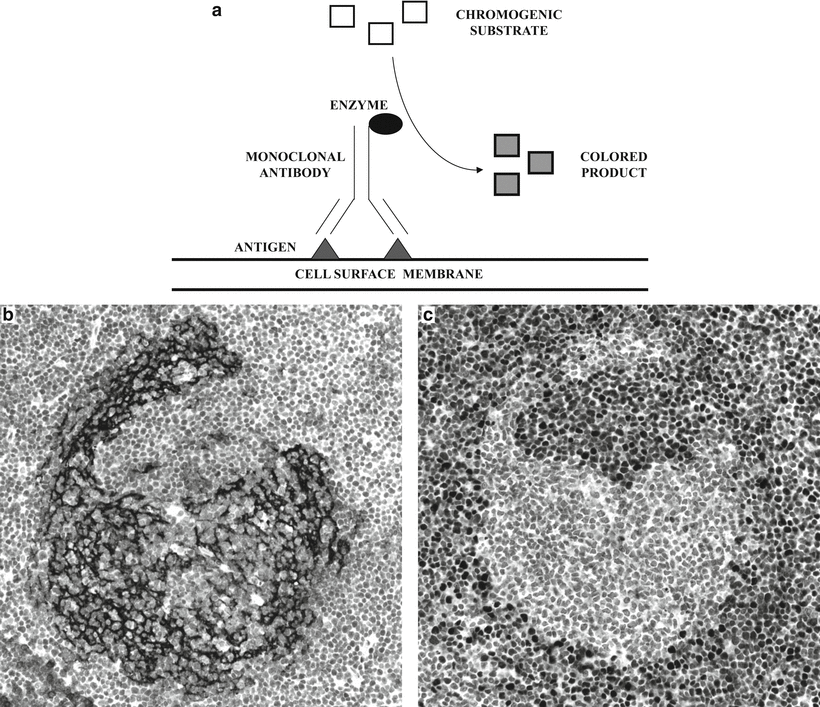
Fig. 31.1
Immunopathological detection of cell surface antigens . (a) Diagram illustrating the basic principle of immunopathological methods. An enzyme is linked to a monoclonal antibody, which recognizes a specific cell surface membrane antigen. A chromogenic substrate is added and is enzymatically converted to a colored product, which is visualized microscopically. (b) Lymph node biopsy with partial disruption of the follicular dendritic cell meshwork (highlighted by antibody to CD23) in a case of mantle cell lymphoma. The mantle cell lymphoma cells are not immunoreactive for CD23. (c) The diagnosis of mantle cell lymphoma is supported by nuclear immunoreactivity for cyclin D1.
This chapter will focus on the underlying pathogenesis of lymphoproliferative disorders with some reference on how that information can be used for diagnostic and prognostic purposes. First, there will be an overview of normal B- and T-cell development, noting that the various stages of lymphocyte development can be roughly correlated with different types of lymphoma. A description of immunoglobulin gene rearrangement and T-cell receptor gene rearrangement is imperative since this process provides the basis for the detection of clonality of B- and T-cells and because it highlights some of the differences in subtypes of lymphomas. Because an in depth discussion of the pathogenesis of lymphoproliferative disorders is enough to fill several textbooks, a select group of non-Hodgkin lymphomas will be reviewed with particular attention to how the pathogenetic mechanisms can be used to classify, diagnose, and in some cases serve as a potential therapeutic targets. Finally, there will be a brief discussion of molecular profiling and microRNAs, two areas in which our understanding of lymphomagenesis stands to grow a great deal in the coming years.
31.2 Physiology of B-Cell and T-Cell Development
31.2.1 Normal B-Cell Development
The B lymphocytes arise from pluripotent stem cells in the bone marrow , which subsequently migrate to secondary lymphoid organs including lymph nodes, spleen, and various mucosal sites (e.g., Waldeyer’s ring, Peyer’s patches). There are two phases of B-cell development: the antigen-independent phase of B-cell differentiation that occurs in the bone marrow, independent of antigen exposure and giving rise to naïve B-cells, and the antigen-dependent phase of differentiation that primarily occurs in the extramedullary compartment and secondary lymphoid organs after exposure to antigen [4, 5]. Definable intermediate stages of the antigen-dependent phase have been characterized, ultimately related to the germinal center reaction in secondary lymphoid organs and proliferation of memory B-cells and immunoglobulin-secreting plasma cells.
An orderly progression of morphologic, molecular, and immunophenotypic changes allows recognition of distinct stages of B-cell development from the progenitor B-cells to the terminally differentiated plasma cells. The stages of B-cell development are illustrated in Fig. 31.2. At the molecular level, the genes coding for the immunoglobulin heavy and light chain proteins undergo sequential rearrangements early in B-cell development (Fig. 31.2). Initially, the immunoglobulin mu heavy chain located on chromosome 14q32 undergoes rearrangement, followed by kappa light chain rearrangement on chromosome 2p12 and lambda light chain rearrangement on chromosome 22q11 [6]. Subsequent transcription and translation of the mu heavy chain gene results in the appearance of cytoplasmic mu heavy chain protein, which defines the pre-B-cell stage of development. Antigen-independent differentiation leads to mature, but naïve B-cells that produce intact surface immunoglobulin molecules (surface IgM and IgD), including two heavy and two light chains (Fig. 31.3a). In the lymph node, the primary antigen response leads to development of B-immunoblasts, which are capable of progressing to IgM-secreting mature B-cells. Several days after antigen exposure, the T-cell-dependent germinal center reaction occurs. This complex reaction involves rapid proliferation of surface Ig-negative centroblasts that have switched off the gene encoding the protein bcl-2, rendering them susceptible to death via apoptosis. In addition, expression of bcl-6 protein and somatic mutation of the bcl-6 and immunoglobulin variable region genes (discussed below) occur. As centroblasts mature to centrocytes, mutations in the immunoglobulin variable region (IgV) occur and expression of surface Ig resumes. Centrocytes whose IgV mutations result in immunoglobulins that recognize self-antigens or show decreased antigen affinity undergo programmed cell death. By contrast, centrocytes whose Ig mutations yield immunoglobulins with increased antigen affinity are saved from apoptosis and resume expression of bcl-2. These veterans of the germinal center reaction gives rise to antigen-specific memory B-cells which can subsequently differentiate into plasma cells.
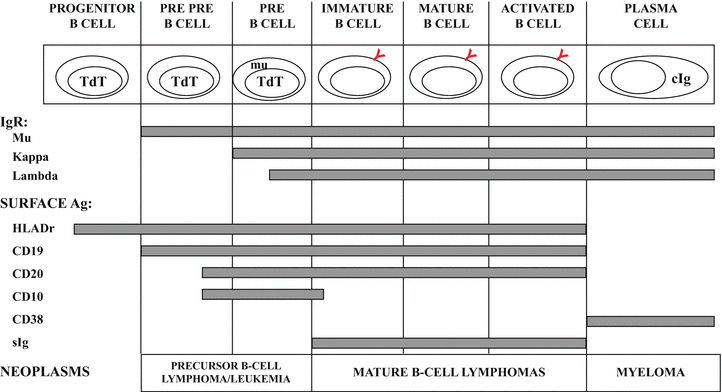
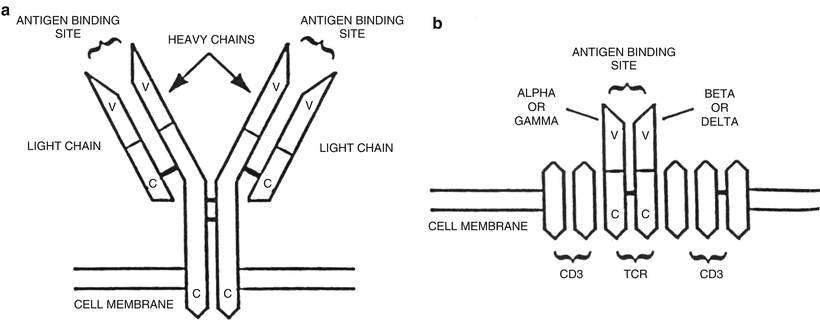
Fig. 31.2
Normal stages of B-cell development with immunological and molecular genetic changes at different stages. See text for discussion. TdT terminal deoxynucleotidyl transferase, mu cytoplasmic mu heavy chain, cIg cytoplasmic immunoglobulin, IgR immunoglobulin rearrangement, sIg surface immunoglobulin.
Fig. 31.3
Schematic diagram of immunoglobulin and T-cell receptors. (a) The immunoglobulin protein is a heterodimer composed of two heavy and two light chains, each of which has variable (V) and constant (C) regions. (b) The T-cell receptor (TCR) is also a heterodimer composed of either one α (alpha) and one β (beta) chain or one γ (gamma) and one δ (delta) chain. Each of the TCR proteins has variable (V) and constant (C) regions. CD3 is a complex of five proteins associated with the TCR.
As illustrated in Fig. 31.2, a variety of cellular antigens can be detected at different stages in normal B-cell development. The CD (cluster designation) numbers have been applied to many of these antigens to standardize the nomenclature and allow easier comparison among studies using various antibody clones that recognize the same surface marker. The earliest antigens identified on precursor cells of B lineage are terminal deoxynucleotidyl transferase (TdT), the progenitor cell antigen CD34, the common acute lymphoblastic antigen (CALLA, CD10) and HLA-Dr. However, none of these antigens are B-lineage specific. Specific B-lineage associated antigens, CD19, CD79a, cytoplasmic CD22 and mu heavy chain, and the nuclear transcription factor, B-cell specific activator protein (BSAP, pax-5) are expressed in the pre-B stage of B-cell development [1, 7]. As B-cells mature, there is loss of CD10 and acquisition of the B-cell antigen, CD20 as well as surface immunoglobulin. CD10 is noteworthy in that its expression resumes for the germinal center stage of development, and at the same time, the anti-apoptosis protein, bcl-2 is lost but returns after the germinal center stage. As a B-cell matures to a terminally differentiated plasma cell, the majority of B-cell surface antigens are no longer expressed and CD138 (syndecan-1) appears [8]. The plasma cell is also characterized by presence of abundant cytoplasmic immunoglobulin.
One fundamental theory of lymphoid neoplasia is that disorders of lymphoid cells represent neoplastic transformation of cells at various stages of normal development [9]. This notion has been used in the classification of lymphoid malignancies [1]. For example, precursor B-cell acute lymphoblastic lymphoma/leukemia immunophenotypically mimics normal precursor B-cells by showing expression for TdT, HLA-Dr, CD10, CD19, variable CD20, and (cytoplasmic) mu heavy chain (Fig. 31.2), although the pattern of antigen expression in neoplastic populations differs from that seen in normal precursor B-cells [10]. The mature B-cell neoplasms represent different stages of B-cell development, including mantle cell lymphoma, which represents a neoplasm of CD5-positive naïve B-cells that have not undergone the germinal center reaction [11] and follicular lymphoma, which has an immunophenotype and molecular profile analogous to B-cells at the germinal center stage of development with expression of CD10, bcl-6, and somatic hypermutation of IgH and bcl-6 genes [12]. Plasma cell myeloma is the neoplastic counterpart of the normal plasma cell.
31.2.2 Normal T-Cell Development
T-lymphocytes also arise from pluripotent stem cells in the bone marrow . However, in contrast to B-cell development in which the earliest stages of maturation occur in the bone marrow, progenitor T-cells migrate from the bone marrow to the thymus where the early stages of T-cell development occur [13, 14]. Subsequently, mature T-cells circulate in the peripheral blood and seed peripheral lymphoid tissues, which include paracortical areas of lymph nodes and periarteriolar lymphoid sheaths of the spleen.
Figure 31.4 shows the normal stages of T-cell development in the thymus, which are analogous to B-cell development and occur in an orderly fashion. T-lymphocytes possess a surface membrane protein complex referred to as the T-cell receptor, which is structurally similar to the B-cell surface immunoglobulin receptor (Fig. 31.3b). The T-cell receptor is a heterodimer protein comprised of either one α (alpha) and one β (beta) polypeptide chain, which combine to form the αβ T-cell receptor, or one γ (gamma) and one δ (delta) polypeptide chain, which combine to form the γδ T-cell receptor [15–18]. Approximately 1–5 % of circulating T-cells expresses the γδ T-cell receptor while the remainder expresses the αβ T-cell receptor [18, 19]. T-cells bearing the γδ receptor comprise a greater percentage of T-lymphocytes at mucosal sites and in the spleen. The genes that code for each of the polypeptide chains that make up the T-cell receptor undergo sequential rearrangements early in T-cell development. The first T-cell receptor gene to rearrange is δ, which is followed sequentially by γ, β, and α genes. The genes coding for the α and δ polypeptide chains are located on chromosome 14q11, the β chain gene on chromosome 7q34 and the γ chain gene on chromosome 7p15 [6].
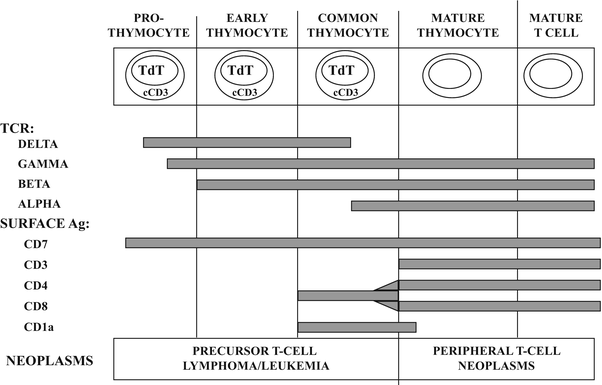
Fig. 31.4
Normal stages of T-cell development with immunological and molecular genetic changes at different stages. See text for discussion. TdT terminal deoxynucleotidyl transferase, cCD3 cytoplasmic CD3, TCR T-cell receptor rearrangements, Ag antigen.
Analogous to developing B-cells, a variety of cellular antigens can be detected at different stages of normal T-cell development (Fig. 31.4). The earliest antigens expressed are TdT and the T-lineage antigen, CD7. The CD3 antigen, which is a protein complex associated with the T-cell receptor (Fig. 31.3b), is primarily a cytoplasmic antigen early in T-cell development and only appears on the cell surface later in development at the same time as the T-cell receptor β chain. The earliest thymocytes lack expression of CD3, CD4, and CD8, but as development progresses, the developing T-cells acquire CD1a, CD2, and CD5. The common thymocyte stage of T-cell development is characterized by coexpression of the CD4 (helper/inducer) and CD8 (cytotoxic/suppressor) T-cell subset antigens. Phenotypically mature T-cells (also referred to as peripheral T-cells) lack TdT, lack CD1a and express surface CD3 with either CD4 or CD8, but not both. After leaving the thymus, naïve T-cells home to the paracortex of lymph nodes and periarteriolar lymphoid sheaths of the spleen. T-cells undergo antigen-dependent differentiation similar to B-cells with evolution through a T-immunoblast stage, giving rise to antigen-specific T-helper cells (CD4 positive), T-suppressor cells (CD8 positive) which show coexpression of either αβ or γδ T-cell receptor [16].
Similar to B-cell non-Hodgkin lymphoma, the T-cell neoplasms represent neoplastic transformation of cells at various stages of normal T-cell development [9] although the correlation is not quite as well characterized as it is for the mature B-cell neoplasms. For example, precursor T-cell lymphoblastic lymphoma/leukemia may exhibit an immunophenotype which resembles the normal common thymocyte, showing expression for TdT, CD1a, cytoplasmic CD3 and CD7, as well as coexpression for CD4 and CD8 (Fig. 31.4). Examples of T-cell non-Hodgkin lymphoma, which are neoplastic counterparts to relatively mature or peripheral T-lymphocytes, include anaplastic large cell lymphoma, cutaneous T-cell lymphoma (mycosis fungoides), and peripheral T-cell lymphoma [1].
31.2.3 B-Cell Surface Immunoglobulin and T-Cell Receptor Gene Rearrangement
The B-cell surface immunoglobulin receptor and T-cell receptor are involved in the process of antigen recognition by normal B-cells and T-cells, respectively. These receptors are structurally similar being heterodimer proteins linked by disulfide bonds and are composed of both variable (V) and constant (C) regions [6] (Fig. 31.3). The variable regions of these proteins are similarly involved in antigen recognition. The constant region of the immunoglobulin heavy chain protein defines the different immunoglobulin subclasses (IgG1, IgG2, IgG3, IgG4, IgA1, IgA2, IgM, IgD, and IgE) [5]. The genes that code for the B-cell and T-cell receptors are also structurally similar and consist of a large number of exons or coding sequences referred to as a supergene family. The exons undergo a process of DNA recombination or rearrangement leading eventually to transcription and translation and the production of functional receptor proteins [5, 6, 15, 18, 20].
A general scheme illustrating the process of B-cell surface immunoglobulin and T-cell receptor gene rearrangement is shown in Fig. 31.5. The germline configuration refers to non-rearranged DNA. The exons which code for the variable regions of the immunoglobulin and T-cell receptors are referred to as variable (V) segments, diversity (D) segments, and junctional (J) segments, and those which code for the constant regions are referred to as (C) segments. The process of gene rearrangement first involves the selective apposition of one D segment with one J segment by deletion of the intervening coding and noncoding DNA sequences resulting in a DJ rearrangement. A similar process of rearrangement opposes a V segment, located in the 5′ direction to D and J to form a VDJ rearrangement. Transcription to messenger RNA (mRNA) then occurs even though the rearranged VDJ segments are not yet directly opposed to C segments, which are remotely located in the 3′ direction. Subsequent splicing of the mRNA with deletion of noncoding sequences results in apposition of VDJ with C to form a VDJC mRNA, which can then be translated into a surface immunoglobulin or T-cell receptor protein.
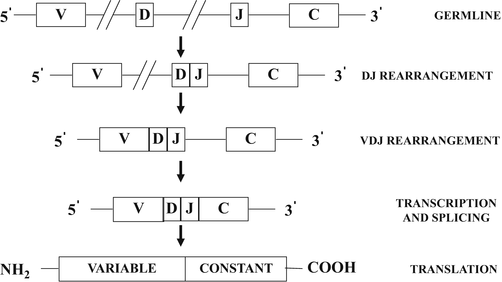
Fig. 31.5
Schematic diagram illustrating the sequential steps involved in immunoglobulin and T-cell receptor gene rearrangements . See text for discussion. V variable segments, D diversity segments, J junctional segments, C constant segments.
The genes coding for the immunoglobulin heavy chain proteins and T-cell receptor beta and delta chain proteins include V, D, J, and C segments. The genes coding for the kappa and lambda light chain proteins and the T-cell receptor alpha and gamma proteins include only V, J, and C segments without D segments [5, 6, 20]. A schematic diagram of the immunoglobulin heavy chain and the T-cell receptor beta chain supergene families is shown in Fig. 31.6. The immunoglobulin heavy chain locus contains 123 VH segments, 27 D segments, 6 JH segments [21]. The VH segments, which include 79 pseudogenes and 44 segments that have an open reading frame, can be further divided into 7 families based on the presence of conserved or homologous DNA sequences, which are common to each member of a family. The VH families are designated VH1 to VH7. The T-cell receptor beta chain gene includes 75–100 Vβ segments, which can be further divided into several families, and two tandem DJC complexes referred to as D1J1C1 and D2J2C2. Each DJC complex contains one D segment and one C segment. The first DJC complex contains six Jβ segments (Jβ1 group) and the second DJC complex contains seven Jβ segments (Jβ2 group) [6, 15, 20].
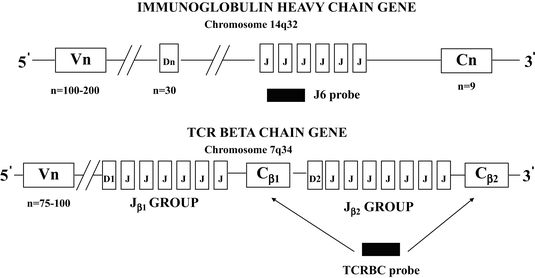
Fig. 31.6
Schematic diagram of the immunoglobulin heavy chain and the T-cell receptor beta chain supergene families. The specific sites of recognition for probes used for Southern blotting are typically associated with consensus sequences in the joining (J6 probe) and constant regions (TCRBC probe).
The humoral (B-cell) and cell mediated (T-cell) systems must be able to recognize a wide variety of environmental antigens even though only a finite quantity of DNA is present in each cell. The complex process of DNA recombination or rearrangement involving the surface immunoglobulin and T-cell receptor allows for tremendous diversity of both the humoral and cell mediated systems, and hence, the ability of these systems to detect a wide variety of antigens [5, 6, 15, 18, 20]. The large number of V, D, J, and C segments results in many combinations, which can be transcribed and translated to millions of different antigen receptors.
Further diversification of the immunoglobulin receptor genes is achieved through a process of somatic hypermutation , which occurs normally within germinal center B-cells. In addition to conserved or homologous DNA sequences within V segments referred to as framework regions (FR), individual immunoglobulin heavy and light chain V segments contain complementarity-determining regions (CDR), which contain nucleotide sequences that encode for the antigen-binding site (Fig. 31.7). The CDRs contain hypervariable nucleotide sequences, which tend to undergo somatic hypermutation. This process results in a series of DNA point mutations with consequent amino acid substitutions within the antigen-binding site. The end product of somatic hypermutation involving the CDRs is enhanced antibody diversity, affinity, and specificity for target antigens [5, 6, 15, 18, 20].
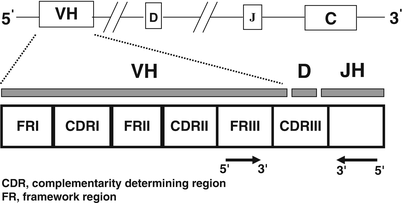
Fig. 31.7
Schematic diagram illustrating the rearranged immunoglobulin heavy chain region. Variable primer sets targeting the complementarity-determining regions or framework regions are used for polymerase chain reaction and southern blot studies to determine B-cell clonality. Arrowheads identify restriction enzyme cleavage sites.
31.3 Molecular Mechanisms in Non-Hodgkin Lymphoma
31.3.1 Background
Non-Hodgkin lymphoma comprises a heterogeneous group of lymphoid neoplasms, which occur because of neoplastic transformation of lymphocytes at different stages of B-cell and T-cell development [4, 13]. The wide variety of B-cell and T-cell non-Hodgkin lymphomas reflects the varying stages of lymphocyte development and the complexity of the immune system. The clinical and pathological characteristics of the non-Hodgkin lymphomas are summarized in a comprehensive manner in the World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues (WHO Classification) [1].
Centrally important to establishing a diagnosis of non-Hodgkin lymphoma is the identification of an abnormal clonal lymphoid population since B-cell and T-cell non-Hodgkin lymphomas represent monoclonal proliferations of B-cells and T-cells, respectively. A major application of molecular genetic methods (e.g., PCR, Southern blot) in the evaluation of non-Hodgkin lymphoma involves the determination of B-cell and T-cell clonality . These methods are considered to be the gold standard for determining clonality but are utilized primarily when a definitive diagnosis (malignant or benign) cannot be determined by morphologic or immunopathologic methods. For B-cell neoplasms, clonality can often be determined immunopathologically by demonstrating the presence of monoclonal surface immunoglobulin [22]. In contrast, for T-cell malignancies, there is no immunopathological equivalent to monoclonal surface immunoglobulin, although qualitative or quantitative loss of normal T-cell antigen expression is considered to be presumptive evidence of T-cell neoplasia [22]. Thus, molecular genetic approaches for the determination of clonality in T-cell non-Hodgkin lymphoma are especially important.
Other uses for molecular genetic techniques in the assessment of lymphoid malignancies include determination of B-cell or T-cell lineage, detection of chromosomal translocations, and detection of minimal residual disease. The latter application has become increasingly useful in evaluating patients before and after bone marrow transplantation particularly in acute lymphoblastic leukemias [23]. The clinical significance of minimal residual disease detection in chronic lymphoproliferative disorders remains a matter of debate and warrants more study [24, 25]. The detection of a specific chromosomal translocation may be very useful in the subclassification of non-Hodgkin lymphoma. For example, in a lymph node with suspected follicular lymphoma, the detection of a translocation, t(14;18), involving the BCL2 proto-oncogene, would confirm this diagnosis. Similarly, in the appropriate context, the detection of a translocation, t(11;14), involving the CCND1 proto-oncogene, would confirm a diagnosis of mantle cell lymphoma or a t(8;14) involving the c-MYC oncogene would support a diagnosis of Burkitt lymphoma [26].
There are many potential mechanisms of carcinogenesis and lymphomagenesis . In some instances, the details of the pathways involved in the transformation of a benign cell to a malignant one are generally well understood, but in most the pathogenesis is a matter of ongoing study. Some diseases can almost be defined by a single, recurrent cytogenetic or molecular alteration that leads to dysregulation of the cell cycle and proliferation of abnormal cells. For example, in mantle cell lymphoma and Burkitt lymphoma, the alteration involves placement of a normally carefully regulated gene that promotes cell growth and division (CCND1 and MYC, respectively) under the regulatory control of the constitutively active immunoglobulin heavy chain gene. In a similar fashion, the common molecular rearrangement in follicular lymphoma puts the normally tightly controlled expression of the BCL2 gene under the control of the immunoglobulin heavy chain gene. However, in this case, the product of the upregulated BCL2 gene does not lead to cell proliferation but rather a blockade of apoptosis, again culminating in an uncontrolled overgrowth of cells. Marginal zone B-cell lymphoma is associated with multiple different translocations that all lead to dysregulation of the NF-kB pathway which plays many roles in cell proliferation and differentiation. The mechanisms involved in the production of functional immunoglobulins (e.g., VDJ recombination, somatic hypermutation, immunoglobulin class switching) provide the molecular milieu for development of many of the familiar translocations in B-cell lymphomas [27].
In contrast, relatively few of the T-cell lymphoproliferative disorders have recognizable recurrent genetic abnormalities. One that does is T-cell prolymphocytic leukemia, which has a mechanism similar to the B-cell lymphoproliferative disorders listed above, placing an oncogene under control of the promoters of the T-cell receptor α/β locus. In the case of anaplastic large cell lymphoma (ALCL), the creation of a novel fusion protein (derived from a translocation involving nucleophosmin (NPM) and anaplastic lymphoma kinase (ALK) genes) with a constitutively active tyrosine kinase domain drives cell proliferation. Another T-cell lymphoma, adult T-cell leukemia/lymphoma (ATLL) has a different mechanism of oncogenesis, being caused by a human retrovirus, HTLV-1.
One thing that has become clear with our understanding of these molecular hallmarks of disease (e.g., IGH/BCL2 rearrangement in follicular lymphoma, NPM/ALK rearrangement in ALCL, HTLV-1 infection in ATLL) is that the presence of the t(14;18), t(2;5), or HTLV-1 infection should be regarded as “required but not sufficient” for malignant transformation. This conclusion must be reached, as many normal subjects can be shown to have molecular evidence of these rearrangements while never manifesting signs of the disease. The discussion of these lymphoproliferative disorders will examine how these molecular alterations can lead to cell cycle disruption and some possible additional hits that may help produce a clinical lymphoma, as well as touch on how the use of molecular diagnostic tests can be used to diagnose and follow these disorders.
31.3.2 Mantle Cell Lymphoma (t(11;14)(q13;q32), GH-CCND1)
Mantle cell lymphoma (MCL) is a pathologically distinct B-cell lymphoproliferative disorder [28] that is characterized by upregulation of cyclin D1, most often due to the presence of a translocation involving the BCL1 (B-cell leukemia/lymphoma-1) region on chromosome 11q13 and the IGH gene on chromosome 14q32. MCL makes up between 5 and 10 % of all B-cell NHL and shows a male predominance. MCL occurs over a wide age range, but is more prominent in older patients with a median age of approximately 60 years. Compared to other B-cell non-Hodgkin lymphomas composed of small lymphocytes, MCL has a poor prognosis with a median survival time of 3–5 years even with aggressive therapy.
Classic cases show effacement of lymph node architecture by a population of small to medium-sized lymphocytes with irregular nuclear contours, evenly dispersed chromatin and scant cytoplasm. A population of nucleolated cells is distinctly absent in typical cases. Scattered mitotic figures and solitary epithelioid macrophages are identified (Fig. 31.8a, b). The neoplastic cells can be distributed in a variety of patterns, including nodular pattern, diffuse pattern, and a mantle zone pattern. Several studies have reported an association between the growth pattern and prognosis [29–31]. Occasional cases of otherwise typical mantle cell lymphoma show moderate to abundant cytoplasm, resembling marginal zone B-cells. Plasma cell differentiation is not typical of mantle cell lymphoma, but has been reported. A more aggressive blastoid variant is characterized by larger cells with more open chromatin, scant cytoplasm, and variably prominent nucleoli (Fig. 31.8c). Leukemic presentations of mantle cell lymphomas also occur, and may present with a marked peripheral blood leukocytosis, raising a differential diagnosis with prolymphocytic leukemia.
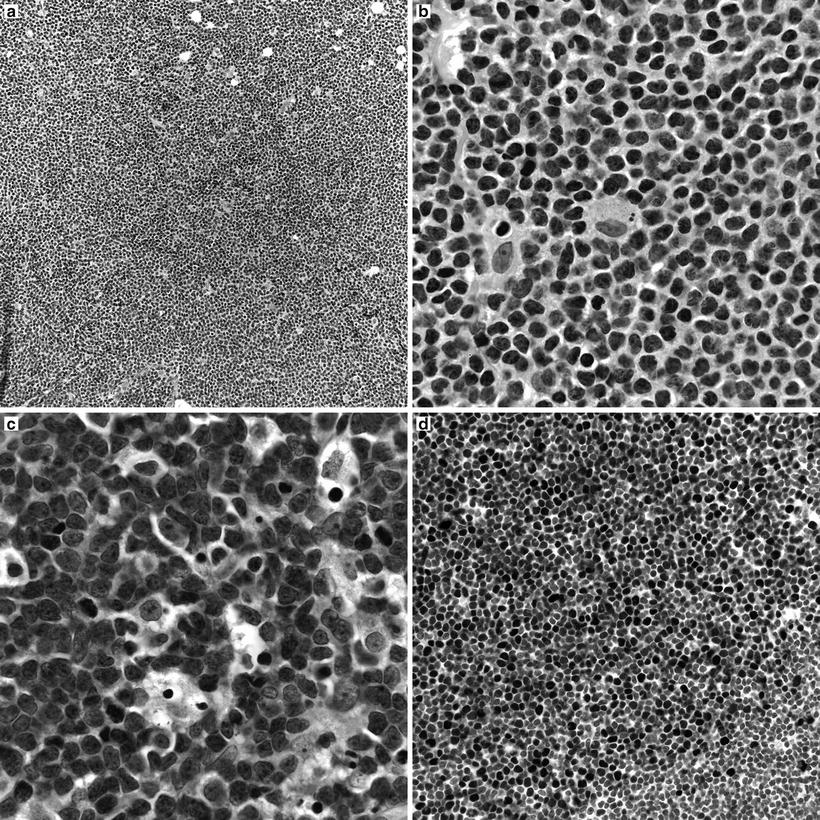
Fig. 31.8
Morpohologic features of mantle cell lymphoma. (a) Low magnification image showing a vaguely nodular pattern of growth. (b) Classical mantle cell lymphoma cytology with small to medium cells showing slightly irregular nuclear contours, inconspicuous nucleoli, and scant cytoplasm. Scattered epithelioid macrophages are present. (c) Blastoid variant of mantle cell lymphoma with larger, nucleolated nuclei and more open chromatin. (d) Cyclin D1 immunohistochemistry highlighting nuclear immunoreactivity in the neoplastic cells.
Flow cytometric immunophenotyping shows that the neoplastic cells in MCL express the B-cell associated antigens CD19, CD20, CD22, and FMC-7. Because this is a lymphoma of mature B-cells, the neoplastic cells express surface immunoglobulins, often IgM and IgD heavy chains and moderate to bright kappa or lambda light chains. One of the phenotypic hallmarks of MCL is coexpression of the T-cell associated antigen, CD5 without coexpression of CD23, CD10, or bcl-6. This immunophenotypic profile supports the notion that mantle cell lymphomas are derived from a population of CD5-positive naïve pre-germinal center B-cells.
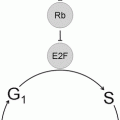
Recognition of t(11;14)(q13;q32) as the genetic hallmark of MCL has greatly improved reproducibility in diagnosis and allowed greater study of this disorder [32]. This translocation involves the BCL1 locus on chromosome 11 and the immunoglobulin heavy chain locus on chromosome 14, with the result that the CCND1 gene (formerly known as PRAD1 because of its association with parathyroid adenoma), which encodes for the protein cyclin D1, comes under regulatory control of the immunoglobulin heavy chain enhancer. This translocation leads to overexpression of the cyclin D1 protein, which is not typically expressed at high levels in B lymphocytes [33]. The cyclin D1 mRNA produced as a result of this translocation has two main forms, each of which contains the complete coding region for a 36 kDa form of cyclin D1: a 4.5 kb form with a long 3′ untranslated region and a 1.5 kb form that lacks the long 3′ untranslated region. Cyclin D1 promotes the transition of cells from the G1 phase into the S phase of the cell cycle by binding to cyclin-dependent kinases 4 and 6 (CDK4 and CDK6). These complexes of cyclin D1 and CDK4 or CDK6 lead to phosphorylation of retinoblastoma 1 (Rb1), eliminating its suppressive effects on the cell cycle by leading to the release of the transcription factor E2F and progression into the S phase. Other molecular pathways are affected including downregulation of the TP53 tumor suppressor gene [34]. Because the t(11;14) with cyclin D1 overexpression is found in virtually all cases, it is felt to be one of the early events in the development of mantle cell lymphoma [35]. However, since it can be seen in normal individuals without MCL and since mouse models with the classic t(11;14) do not develop mantle cell lymphoma without other genes also being affected, it is also felt that this is not sufficient for full transformation. Additional genetic hits are common in MCL, and cytogenetic studies have demonstrated that MCL has one of the highest levels of genetic instability among lymphoproliferative disorders. Many of the additional genetic abnormalities seen in mantle cell lymphoma seem to target either cell cycle regulation (e.g., through deletions of chromosome 9p21 resulting in the loss of two genes, the CDK inhibitor p16INK4a (interferes with Rb1 function) and p14ARF (downregulates the p53 pathway)) or the DNA damage response (mutations of the ATM (ataxia-telangiectasia mutated) gene, which has a role in the cellular response to DNA damage) [33, 34] (Fig. 31.9). Better understanding of the molecular mechanisms leading to mantle cell lymphoma may lead to therapeutic agents that directly target these various pathways [36].
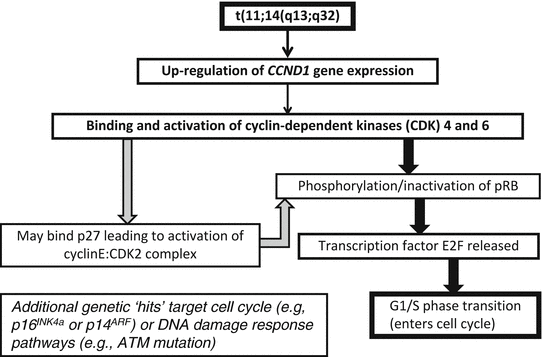
Fig. 31.9
Molecular mechanism in mantle cell lymphoma. The t(11;14)(q13;q32) leads to overexpression of cyclin D1 (CCND1), which binds to cyclin-dependent kinases (CDK) 4 and 6. This leads to over-phosphorylation of pRB, releasing the transcription of E2F and progression from the G1 to S phases of the cell cycle. Deregulation of pRB via other cyclins is also possible. Subsequent genetic abnormalities target the cell cycle and DNA repair pathways.
Some groups have asserted that cyclin D1 positivity should be a part of the definition of mantle cell lymphoma [37]. However, by gene expression profiling, a small subset of cyclin D1-negative mantle cell lymphomas has been identified, which have the usual morphologic features and unique gene expression signature of mantle cell lymphoma [38]. This report suggests that other cyclin D proteins may substitute for cyclin D1 in lymphomagenesis in such cases. Like cyclin D1, other members of the cyclin D family of proteins are involved in cell cycle control, particularly as regulators of progression from the G1 to S phase of the cell cycle.
The diagnosis of MCL is based on a combination of morphologic immunophenotypic, cytogenetic, and clinical data. Identification of cyclin D1 overexpression by immunohistochemical analysis (Fig. 31.8d) has been shown to be a specific and sensitive marker for MCL [39]. Immunohistochemical markers for proliferation (especially Ki-67) have been shown to be of prognostic value [29, 40], a finding shown to be associated with a distinct genetic signature [41]. Classical cytogenetic studies allow identification of the t(11;14) (Fig. 31.10a). Since the specific breakpoints in the BCL1 region are spread over a wide area (Fig. 31.10b), PCR analysis for a specific breakpoint is not as sensitive as other techniques in detecting the rearrangements. FISH analysis has been shown to be a very sensitive marker for detection of the t(11;14) in MCL [42]. Again, it should be borne in mind that while detection of cyclin D1 abnormalities is quite sensitive for mantle cell lymphomas, there seems to be a subset of MCL cases that lack a t(11;14) and cyclin D1 overexpression [38]. It should also be noted that these findings are not entirely specific to MCL) . For example, cyclin D1 overexpression by immunohistochemical analysis is seen in hairy cell leukemia, and some cases of plasma cell myeloma not only show overexpression of cyclin D1 by immunohistochemical analysis but even exhibit an identical t(11;14).
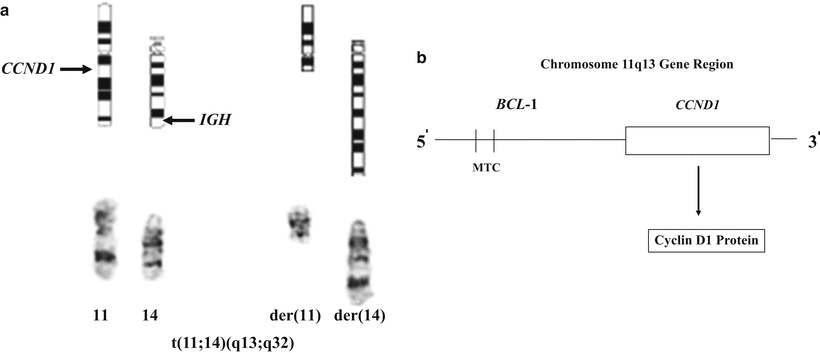
Fig. 31.10




Cytogenetics of mantle cell lymphoma. (a) Derivative chromosomes produced by the t(11;14) translocation (Giemsa-stained chromosomes and ideogram). (b) Schematic diagram of the chromosome 11q13 gene region, which includes the BCL1 locus and the CCND1 gene. The breaks in the 11q13 gene region are widely scattered within the BCL1 locus; however, approximately 30–40 % of breaks occur within the MTC (major translocation cluster) region of the BCL1 locus. The CCND1 gene encodes for the cyclin D1 protein.
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
