Molecular Genetics of Tumors of Thyroid Follicular Cells
James A. Fagin
Yuri Efimovich Nikiforov
Thyroid follicular cells give rise to tumors with distinct pathologic and clinical features. Follicular cell-derived cancers are represented by well-differentiated papillary carcinoma and follicular carcinoma, poorly differentiated carcinoma (also known as insular carcinoma), and anaplastic (undifferentiated) carcinoma. Follicular adenoma is a benign tumor that may serve as a precursor for some follicular carcinomas. Less-differentiated thyroid cancers, i.e. poorly differentiated carcinoma and anaplastic carcinoma, can develop de novo, although many of them likely arise through the process of step-wise dedifferentiation of papillary and follicular carcinomas.
Benign and malignant thyroid neoplasms consist of clonal cell populations (1,2,3,4,5). This indicates their derivation from single precursor cells, with the initial step in tumor development involving the acquisition of a somatic mutation that alters the structure of a gene that confers cells with a growth advantage. Many of the genes involved in the pathogenesis of thyroid tumors have been identified, and found to be preferentially
associated with certain tumor types. Cell proliferation is stimulated by extracellular growth factors that activate specific signaling cascades that dictate the orderly sequence of events needed for DNA synthesis and cell division. Molecules acting at multiple steps along growth signaling pathways can function as oncogenes when their structure is disrupted. In thyroid cancer, many oncogenic mutations lead to the activation of the mitogen-activated protein kinase (MAPK) and PI3K signaling pathways, which are crucial for tumor initiation and progression. In addition, loss-of-function mutations of genes encoding growth inhibitory proteins involved in signaling pathways, cell cycle checkpoints or cell survival contribute significantly to tumorigenesis. Common mechanisms of gene mutations in cancer are point mutations and chromosomal rearrangements. In thyroid cancer, each of these mutational mechanisms appears to be linked to specific etiologic factors.
associated with certain tumor types. Cell proliferation is stimulated by extracellular growth factors that activate specific signaling cascades that dictate the orderly sequence of events needed for DNA synthesis and cell division. Molecules acting at multiple steps along growth signaling pathways can function as oncogenes when their structure is disrupted. In thyroid cancer, many oncogenic mutations lead to the activation of the mitogen-activated protein kinase (MAPK) and PI3K signaling pathways, which are crucial for tumor initiation and progression. In addition, loss-of-function mutations of genes encoding growth inhibitory proteins involved in signaling pathways, cell cycle checkpoints or cell survival contribute significantly to tumorigenesis. Common mechanisms of gene mutations in cancer are point mutations and chromosomal rearrangements. In thyroid cancer, each of these mutational mechanisms appears to be linked to specific etiologic factors.
In addition to somatic mutations, thyroid cancer development involves global alteration in gene expression patterns, dysregulation of microRNAs, and aberrant gene methylation. Among these alterations, somatic mutations represent early events that initiate and drive the transformation process, have been studied most extensively, and represent the primary focus of this chapter.
Growth Factors and Eicosanoids as Thyroid Tumor Progression Factors and Promoters of Angiogenesis
The abundance of TSH in plasma is an important determinant of the mitotic rate of thyroid cells (6). High TSH levels promote progression of certain thyroid neoplasms, including many thyroid cancers. Benign thyroid nodules are a well-known risk factor for thyroid cancer, and several epidemiologic studies have found an association between higher serum TSH levels and risk of thyroid cancer in patients with thyroid nodules (7,8,9). The risk of cancer increases incrementally with higher levels of TSH, even within the normal range. In addition to stimulating cell proliferation, recent experimental evidence in gene targeted mice suggests that oncogenic BRAF activation requires simultaneous TSH stimulation to transform thyroid cells and initiate tumor development (10).
The role of growth factors present in the tumor environment is less clear. Whereas growth of normal cells is highly dependent on external mitogenic stimulation by growth factors, a classical feature of cancer cells is that they are to large extent released from this constraint. Despite this, tumor cells retain variable degrees of responsiveness to polypeptide growth factors, and these may contribute to tumor progression or development. Fibroblast growth factors (FGF) 1 and 2 are mitogens for thyroid follicular cells (11), and are overexpressed in multinodular goiters as well as benign and malignant thyroid neoplasms (12,13,14). TSH requires the presence of insulin-like growth factor I (IGF-I) to exert its full mitogenic effect in human thyroid cells. Moreover, overexpression of IGF-I and the IGF-I receptor in thyroid cells of transgenic mice induces goiter formation (15). Both IGF-I and IGF-II are overexpressed in human thyroid cancers (16,17). Whereas IGF-I is primarily produced by cells in the tumor stroma (18), thyroid cancer cells are believed to be the source of local IGF-II production (16). TGFα (and its close relative EGF) is also a mitogen for thyroid follicular cells in vitro, and is locally expressed within the thyroid (19,20). The mechanism by which tumor cells overexpress growth factors does not involve primary modifications of the growth factor genes themselves. Rather, growth factor overexpression is secondary to oncogenic activation of other signaling pathways. For example, mutations of RAS result in increased TGFα gene expression in rat thyroid FRTL-5 cells (21), and of IGF-I in human thyroid cells (22).
For cancer clones to progress, tumor cells promote growth of capillaries and larger vessels from adjacent tissue in order to deliver nutrients and oxygen and eliminate metabolic waste. Some of the most prominent signals driving the process of angiogenesis have been identified in other cancer types (23,24), and are likely to play similar roles in the thyroid. Vascular endothelial cell growth factors (VEGF) are a family of at least five structurally related polypeptides, VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PIGF), which modulate blood and lymphatic vessel growth (25). VEGF-A is expressed at higher levels in thyroid cancers than in normal thyroid cells or benign thyroid tumors (26,27). VEGF-C, which primarily stimulates lymphangiogenesis, is also overexpressed in thyroid cancers, although this alone is unlikely to account for the predisposition to undergo lymphatic spread, since not all thyroid tumor types prone to lymph node metastasis overexpress this growth factor. Besides its mitogenic effects on thyroid cells, FGF 2 is also a potent angiogenic factor (28). TGFβ, despite inhibiting thyroid cell growth and differentiated function (29), has been implicated in pathogenesis of multinodular goiters (30) and could play a role in vascular remodeling. It is important to consider the role of stromal derived growth factors in tumor evolution, as illustrated by recent evidence indicating that modulation of TGFβ signaling in stromal fibroblasts can impact the oncogenic potential of adjacent epithelial cells (31). In addition to polypeptide growth factors, other secreted products present in the tumor microenvironment can also promote neovascularization. Thyroid cancers, like other epithelial malignancies, have higher expression of cyclooxygenase 2 (COX-2), the enzyme catalyzing the initial step in prostanoid biosynthesis from arachidonic acid (32,33). Tumor derived prostanoid production is a significant contributor to colorectal tumorigenesis in animals and humans (34,35,36). PGE2, the major product of COX-2 acting via microsomal prostaglandin E synthase (mPGES), has been implicated as a key mediator of these effects. Among other properties, PGE2 is a powerful stimulus for angiogenesis. It has been shown that expression of RET or RAS mutants induces PGE2 production via increased expression of COX-2 and mPGES1 in rat thyroid cells, thus providing a link between oncogenic tumor initiation, activation of prostaglandin biosynthesis, and tumor neovascularization (37).
Table 50B.1 Characteristics of different types of RET/PTC rearrangement in papillary thyroid carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mutations Affecting Signal Reception from Extracellular Ligands
Tyrosine Kinase Receptors
Receptor tyrosine kinases (RTK) are common targets of oncogenesis. This can occur through gene amplification of a tyrosine kinase receptor of normal structure and through activating
mutations. For example, the epidermal growth factor receptor (EGFR or Erb1) is overexpressed in certain epithelial malignancies and in glioblastomas, and its close relative Erb-B2/neu is amplified and overexpressed in breast and ovarian cancer (38,39,40). Gene copy number gains of RTK genes such as EGFR, the platelet-derived growth factor receptors (PDGFR) α and β, VEGFR1 and 2 and KIT (41) have been reported in anaplastic and follicular thyroid cancers.
mutations. For example, the epidermal growth factor receptor (EGFR or Erb1) is overexpressed in certain epithelial malignancies and in glioblastomas, and its close relative Erb-B2/neu is amplified and overexpressed in breast and ovarian cancer (38,39,40). Gene copy number gains of RTK genes such as EGFR, the platelet-derived growth factor receptors (PDGFR) α and β, VEGFR1 and 2 and KIT (41) have been reported in anaplastic and follicular thyroid cancers.
Even though in most cases these receptors are structurally intact, their increased abundance could favor higher amplitude of signaling through effector pathways that can promote tumor progression. Indeed, all members of the EGFR family have been reported to be overexpressed in papillary thyroid cancers (42,43). Overexpression of MET, the receptor for hepatocyte growth factor (HGF) is also highly prevalent in these cancers (44,45). This is not due to gene amplification, but may instead be secondary to induction of gene expression, possibly as a result of illegitimate signaling from mutant RAS or RET/PTC oncogenes (46,47). Although a predicted increase in responsiveness to the respective ligands for these tyrosine kinase receptors may participate in the tumorigenic process, this has not been demonstrated.
Growth factor receptors can also lead to cell transformation when their structure is modified through activating mutations. The most significant example relevant to the pathogenesis of papillary thyroid carcinomas are the RET/PTC and TRK oncogenes, which are mutant forms of the tyrosine kinase receptors RET and NTRK1. Of note, a recent study reported the presence of activating EGFR mutations in 30% of papillary thyroid carcinomas from Japan (48), although a subsequent observation based on a much larger series of papillary cancers from Japan and US disputed this finding, reporting no EGFR mutations in these tumors (49).
RET/PTC Rearrangements
The RET gene encodes a transmembrane RTK whose expression is normally restricted to a subset of neuronal and neuroendocrine cells, and to the collecting duct of the kidney. RET is a receptor for growth factors belonging to the glial cell-derived neurotropic growth factor (GDNF) family, which also include neurturin, persephin and artemin (50). The RET protein is not normally present in thyroid follicular cells. Chromosomal rearrangements linking the promoter and N terminal domains of unrelated gene/s to the C-terminal fragment of RET result in the aberrant production of a chimeric form of the receptor in thyroid cells. Rearrangements of RET resulting in its constitutive activation are believed to play a causative role in the pathogenesis of a significant proportion of papillary thyroid carcinomas (51).
Several types of RET/PTC have been identified that differ according to the 5′ partner gene involved in the rearrangement (Table 50B.1). The two most common rearrangement types are RET/PTC1 and RET/PTC3, where RET is fused to ether CCDC6 (H4) or the NCOA4 (ELE1) gene, respectively (52,53). Both these rearrangements are paracentric intrachromosomal inversions, since all fusion partners reside on the long arm of chromosome 10 (54,55). In contrast, RET/PTC2 and nine more types of RET/PTC are all interchromosomal rearrangements formed by RET fusion to genes located on different chromosomes (56,57,58,59,60,61,62,63,64). The fusion proteins generated by these recombinant genes homodimerize in a ligand-independent manner through the action of coiled-coil motifs in the N-termini donated by the respective upstream fusion partners (65), resulting in constitutive activation of the tyrosine kinase domain of RET. RET/PTC transforms thyroid cells in culture (66) and leads to the development of papillary carcinomas in transgenic mice, which has been shown in animals with thyroid-specific expression of RET/PTC1 and RET/PTC3 (67,68,69). There is a high prevalence of RET/PTC expression in occult or microscopic papillary carcinoma (70). These point to RET/PTC as one of the key first steps in thyroid cancer pathogenesis.
The prevalence and specificity of RET/PTC rearrangement varies considerably in the reported series (71,72). This is due in part to a true difference in the prevalence of this alteration in specific age groups and in individuals exposed to ionizing radiation, as discussed later in this chapter. However, in most cases, this is due to heterogeneous distribution of this rearrangement within the tumor and the variable sensitivity of the detection methods. This rearrangement can be present in a significant proportion of tumor cells, in which case it can be detected by multiple methods (clonal RET/PTC) or occur in a small
fraction or single cells within the lesion and be detectable only by ultrasensitive detection techniques (non-clonal RET/PTC) (73,74). Clonal RET/PTC occur in 10% to 20% of papillary thyroid carcinomas and is specific for this tumor type (74,75), whereas non-clonal rearrangements have been reported with a significantly higher prevalence in papillary carcinomas and in a variety of other thyroid tumors and benign lesions [reviewed in Ref. (71)].
fraction or single cells within the lesion and be detectable only by ultrasensitive detection techniques (non-clonal RET/PTC) (73,74). Clonal RET/PTC occur in 10% to 20% of papillary thyroid carcinomas and is specific for this tumor type (74,75), whereas non-clonal rearrangements have been reported with a significantly higher prevalence in papillary carcinomas and in a variety of other thyroid tumors and benign lesions [reviewed in Ref. (71)].
This rearrangement, particularly the RET/PTC1 type, is typically found in papillary carcinomas with classic papillary architecture (76) but can also be seen in other microscopic variants of papillary carcinoma (77,78,79). In tumors arising after radiation exposure, RET/PTC1 was associated with classic papillary histology, and RET/PTC3 with the solid variant of papillary carcinoma (80). Correlation between RET/PTC rearrangement and prognosis in human papillary carcinomas remains incompletely defined. RET/PTC1 rearrangement has been associated with more favorable behavior of papillary carcinomas. Thus, in contrast to papillary carcinomas carrying BRAF or RAS mutations, tumors harboring RET/PTC1 have a very low probability of progression to poorly differentiated and anaplastic carcinomas (78,81,82). However, there is some evidence that tumors carrying RET/PTC3 rearrangement may be prone to dedifferentiation and more aggressive behavior (83,84).
TRK Rearrangement
Somatic rearrangements of another member of the tyrosine kinase receptor gene family, the proto-oncogene NTRK1, which codes for the receptor for nerve growth factor (NGF), are also seen in papillary thyroid cancers, albeit with a far lower prevalence (85). Several thyroid-specific TRK oncogenes, named TRK, TRK-T1, TRK-T2, and TRK-T3, have been isolated (86,87,88). All contain a variable portion of NTRK1, including the tyrosine kinase domain, and differ in the 5′-upstream sequences, contributed by the tropomyosin (TPM3), translocated promoter region (TPR), or TRK-fused (TFG) genes, respectively. Targeted overexpression of TRK-T1 induces lesions resembling PTC in transgenic mice, supporting a role for this oncogene as an alternative pathway in tumor initiation (89).
TRK rearrangements have been reported in up to 10% to 15% of papillary thyroid carcinomas (90,91,92), although in most populations its prevalence is likely to be less than 5%. They can occur in tumors with classic papillary architecture and in different microscopic variants, including papillary microcarcinomas and the follicular and tall cell variants. All three types of the rearrangement are found with approximately similar frequency, and several tumors with still uncharacterized TRK fusions have been reported (91,92).
G-protein Coupled Receptors
Growth of pituitary, thyroid and adrenal cells is controlled in part through polypeptide ligands that activate G protein-coupled receptors of the seven-transmembrane domain family, and that signal in part through stimulation of adenylyl cyclase activity. The TSH receptor (TSHR) can be activated by point mutations leading to aminoacid substitutions that either abrogate the requirement for ligand or enhance responses to ligand-mediated activation (93). Germline mutations of this receptor are associated with syndromes of familial hyperthyroidism (94). More relevant to neoplasia, somatic mutations of the TSHR are observed in many autonomously functioning thyroid nodules (AFTN). These benign tumors are characterized by progressive growth and the ability to synthesize hormones in the absence of TSH stimulation. Activating mutations of TSHR result in aminoacid substitutions at multiple sites of the receptor protein, including the third, sixth and seventh transmembrane domains, and the first, second and third intracellular loops (95). Mutations of TSHR are believed to release the receptor protein from structural constraints present in its unliganded form, thus allowing it to activate Gsα in a constitutive manner (96). Although there is general agreement that TSHR mutations play an important role in pathogenesis of AFTN, there is some debate about the actual prevalence of these abnormalities (97,98). About 50% to 80% of solitary AFTN have TSHR mutations, whereas in autonomous nodules present in multinodular goiters the frequency appears to be less (99). The consensus from clinical studies is that AFTN have a low probability of malignant transformation. Accordingly, activating point mutations of TSHR are quite rare in differentiated thyroid carcinomas (100,101,102,103,104,105,106).
Nuclear Receptors
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that bind to DNA as heterodimers with the retinoid X receptors. The activity of PPARγ is regulated by binding to small lipophilic ligands, mainly fatty acids derived from nutritional intake or metabolism (107). PPARγ has been shown to play an important role in regulating genes involved adipocyte differentiation and lipid metabolism. An involvement of this nuclear receptor in thyroid cancer was discovered by the characterization of a translocation in a subset of human thyroid follicular carcinomas, t(2;3) (q13;p25), which results in fusion of the DNA binding domains of the thyroid transcription factor PAX8 to domains A to F of PPARγ1 (108). PAX8/PPARγ mRNA and protein are present in follicular-pattern thyroid neoplasms. It is found in approximately 35% (range, 0% to 78%) of conventional follicular carcinomas and in less than 5% of oncocytic carcinomas (109). This rearrangement can also be seen in a small proportion (1% to 5%) of the follicular variant papillary carcinomas and in some (∼8%) of follicular adenomas (109,110,111,112,113,114). PAX8/PPARγ-positive tumors tend to present in patients at a younger age, be smaller, and more often show a solid growth pattern and vascular invasion histologically as compared to follicular carcinomas negative for this rearrangement (110,112). Tumors with PAX8/PPARγ almost never show RAS mutations, suggesting that follicular carcinomas develop via two distinct molecular pathways, associated with either PAX8/PPARγ or RAS mutation (112).
The PAX8/PPARγ fusion results in strong overexpression of the PPARγ gene. However, the mechanisms of cell transformation induced by this rearrangement are not fully understood. Several proposed possibilities, some of which contradict each other, include inhibition of normal PPARγ function by the chimeric PAX8/PPARγ protein through a dominant negative effect, activation of normal PPARγ targets due to the overexpression of the chimeric protein that contain all functional
domains of wild-type PPARγ, deregulation of PAX8 function, and activation of a set of genes unrelated to both wild-type PPARγ and wild-type PAX8 pathways (108,115,116). In some experimental models a paradoxical tumor suppressive property of this chimeric protein by inhibition of tumor angiogenesis was noted (117,118). Thyroid-specific overexpression of PAX8/PPARγ in transgenic mice results in mild hyperplasia after one year, suggesting that other cooperating events are likely required for transformation (119).
domains of wild-type PPARγ, deregulation of PAX8 function, and activation of a set of genes unrelated to both wild-type PPARγ and wild-type PAX8 pathways (108,115,116). In some experimental models a paradoxical tumor suppressive property of this chimeric protein by inhibition of tumor angiogenesis was noted (117,118). Thyroid-specific overexpression of PAX8/PPARγ in transgenic mice results in mild hyperplasia after one year, suggesting that other cooperating events are likely required for transformation (119).
In addition to PAX8/PPARγ, the PPARγ gene can be fused in thyroid tumor cells to other partners. A novel fusion partner is CREB3L2. This type of PPARγ fusion is rare and has been reported in one series in 1 out of 42 (2.4%) follicular carcinomas (120).
A remarkably high prevalence (>90%) of mutant thyroid hormone receptor (TR) α1 and β1 transcripts was reported in human papillary thyroid carcinomas in one study (121). The mutations reported in TR were predicted to have impaired transactivation properties, and to act in a dominant negative fashion. However, these findings were not confirmed in a separate independent study, in which no such mutations were found despite full length sequencing of TRβ1 cDNAs from a similar number of thyroid cancers (122). Homozygous gene targeted mice with a mutant TRβ (TRβPV mice) develop invasive and metastatic thyroid cancers (123). Although this is consistent with a role for TRβ in thyroid cancer progression, these mice have extreme thyroid hormone resistance and very high TSH levels throughout life. Subsequent studies have shown that TSH is required but is not sufficient to induce tumorigenesis in this mouse model. Recent evidence suggests that the tumorigenic process may be favored by non-genomic effects of thyroid hormone (124,125), possibly mediated via phosphinositol-3 kinase (PI3K)–AKT–mTOR signaling (126). Hence, although loss-of-function of TR has not been convincingly demonstrated in human thyroid cancers, the insights from mouse models of generalized thyroid hormone resistance have unmasked intriguing non-canonical effects of thyroid hormone on thyroid cells that may play a role in carcinogenesis.
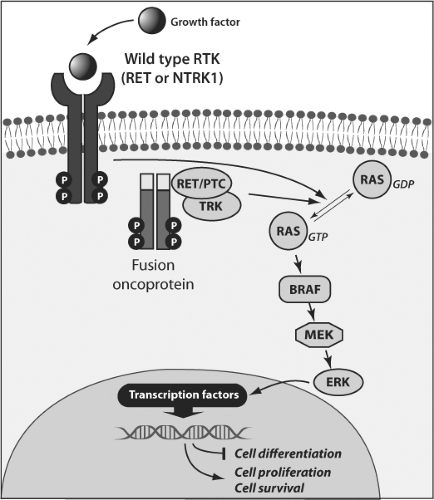 Figure 50B.1 Schematic representation of the MAPK signaling pathway and its activation in thyroid cancer. Binding of growth factors to wild-type receptor tyrosine kinases (RTK), such RET and NTRK1, results in receptor dimerization and activation via autophosphorylation of tyrosine residues in the intracellular domain. In thyroid cancer, the receptors are activated constitutively because the kinase domain is fused to N-terminal proteins that dimerize in a ligand-independent manner. The activated receptor through a series of adaptor proteins leads to activation of RAS located at the inner surface of the plasma membrane by substitution of GDP with GTP. The active, GTP-bound form of RAS binds to and recruits RAF proteins, mainly BRAF in thyroid follicular cells, to the plasma membrane. Activated BRAF phosphorylates and activates MEK and ERK. Once activated, ERK phosphorylates cytoplasmic proteins and translocates into the nucleus, where it regulates transcription of the genes involved in thyroid hormone biosynthesis, proliferation, and survival. Activation of this pathway in thyroid cancer occurs as a result of RET/PTC and TRK chromosomal rearrangements or point mutation affecting the RAS and BRAF genes. |
Defects in the Intracellular Transmission of the Signal
RAS Oncogenes
After ligand binding, tyrosine kinase receptors dimerize, and become autophosphorylated on selected tyrosine residues within their cytoplasmic region. The phosphorylated tyrosines create high affinity binding sites for proteins containing Src homology 2 (SH2) domains. Recruitment of SH2-containing proteins represents the initial link in a chain of phosphorylation reactions that relay and amplify the growth factor-receptor signal. Coupling to Ras takes place through the SH2/SH3 adaptor protein Grb2, which is in turn linked to Sos proteins that activate Ras by exchange of GDP for GTP. GTP-bound RAS is inactivated by hydrolysis of its associated GTP to GDP, which is dependent on its intrinsic GTPase activity, and on interaction with proteins (GTPase-activating proteins or GAPs) that markedly increase the hydrolysis of Ras-bound GTP. Three RAS genes are prominent in cancer pathogenesis: H-RAS, K-RAS, and N-RAS. From a biochemical standpoint their properties are quite similar, and all can be converted to an oncogenic form. Point mutations in discrete domains within RAS that increase protein affinity for GTP, or inactivate its GTPase function, permanently switch the protein to the “on” position. Activation of Ras leads to the activation of MAP kinases, thus linking the activity of cell surface receptors to critical intracellular targets (Fig. 50B.1). MAP kinase activity is regulated through the sequential activation of three kinases, which in the case of extracellular-signal-related kinases (ERKs) includes successive activation of RAF and MEK kinases. As discussed in greater detail below, this pathway is believed to be central to RAS-mediated thyroid dedifferentiation and transformation (127,128).
RAS mutations are found in all types of thyroid follicular cell-derived tumors (prevalence is N-RAS > H-RAS > K-RAS). RAS mutations are also found in 40% to 50% of conventional type follicular carcinomas (129,130,131,132,133,134) and 20% to 40% of conventional type follicular adenomas (129,130,131,132,135). A lower incidence has been seen in oncocytic tumors (136,137). In papillary thyroid carcinomas, RAS mutations occur in 10% to 20% of cases (134,135,138,139,140,141). Virtually all papillary carcinomas harboring RAS mutation have the follicular variant histology (76,142). Some early studies reported RAS mutations in hyperplastic nodules. However, since these lesions carry a clonal mutation, they represent neoplasms and therefore should be designated as follicular adenomas.
The fact that RAS mutations are present in both benign and malignant thyroid neoplasms suggests that RAS activation may be an early step in thyroid tumor development (129,143). In support of this, transgenic mice with thyroid-specific expression of mutant K-RAS and N-RAS develop benign thyroid lesions (144,145,146). However, knock-in mice expressing endogenous levels of oncogenic mutants of KRAS or HRAS do not develop thyroid lesions, indicating that somatic activation of single RAS alleles is insufficient to drive tumorigenesis (147,148) whereas development of malignant tumors requires either additional prolonged goitrogen stimulation or loss of PTEN (147).
Some studies have found that RAS mutations in thyroid carcinomas correlate with tumor dedifferentiation and less favorable prognosis (134,149). Several studies have found a significant correlation between RAS mutation and metastatic behavior of follicular and papillary carcinomas, especially with respect to bone metastases (134,138,139,149,150). On the other hand, RAS mutations are frequently found in encapsulated follicular variants of papillary carcinoma, a tumor with an indolent behavior (142,151). Therefore, it is likely that RAS mutations mark a subset of well-differentiated thyroid carcinomas which are prone to metastatic spread and dedifferentiation but this mutation cannot be used as a universal prognostic marker for all types of thyroid tumors.
The functional consequences of mutagenic RAS activation in thyrocytes are only partially understood. Activation of RAS induces cell proliferation in primary cultures of human thyrocytes (152,153) and in rat thyroid cell lines (154,155). In human thyroid cells, RAS-induced proliferation is self-limited, through a mechanism that may require reactivation of expression of the cyclin-dependent kinase inhibitor p16(INK4a) (152,156). By contrast, in immortalized rat thyroid cell lines, RAS-induced cell proliferation is followed by genomic instability (154,157) and apoptosis (155,158). Mutant RAS is not
by itself sufficient to induce thyroid cell transformation, as determined in gene-targeted mice (147,148) and by inability to form tumors in athymic mice or colonies in soft agar (159). In primary cultures of human thyroid cells, RAS activation does not impair thyroid differentiated function, whereas in immortalized rat thyroid cell lines, overexpression of mutant RAS inhibits TSH-induced iodine uptake, and expression of thyroglobulin (TG), the sodium iodide symporter (NIS) and thyroid peroxidase (TPO) (160) through a mechanism requiring RAF-MEK-ERK signaling (128). Interestingly, when RAS oncoproteins are expressed at physiologic levels in gene targeted mice there is no impairment in thyroid gene expression or thyroid hormone biosynthesis (148,149) in contrast to endogenous expression of oncogenic BRAF in analogous murine models (10,161). Taken together, these data suggest that in human thyroid cancers harboring RAS mutations, loss of differentiated gene expression, where present, likely requires cooperating events that augment RAS signaling, primarily via the MAP kinase pathway.
by itself sufficient to induce thyroid cell transformation, as determined in gene-targeted mice (147,148) and by inability to form tumors in athymic mice or colonies in soft agar (159). In primary cultures of human thyroid cells, RAS activation does not impair thyroid differentiated function, whereas in immortalized rat thyroid cell lines, overexpression of mutant RAS inhibits TSH-induced iodine uptake, and expression of thyroglobulin (TG), the sodium iodide symporter (NIS) and thyroid peroxidase (TPO) (160) through a mechanism requiring RAF-MEK-ERK signaling (128). Interestingly, when RAS oncoproteins are expressed at physiologic levels in gene targeted mice there is no impairment in thyroid gene expression or thyroid hormone biosynthesis (148,149) in contrast to endogenous expression of oncogenic BRAF in analogous murine models (10,161). Taken together, these data suggest that in human thyroid cancers harboring RAS mutations, loss of differentiated gene expression, where present, likely requires cooperating events that augment RAS signaling, primarily via the MAP kinase pathway.
BRAF
There are three isoforms of the serine-threonine kinase RAF in mammalian cells: ARAF, BRAF, and CRAF, with different tissue distribution of expression (162). Although all RAF isoforms activate MEK phosphorylation, they are differentially activated by oncogenic RAS. In addition, BRAF has higher affinity for MEK1 and MEK2 and is more efficient in phosphorylating MEKs than other RAF isoforms (163). The most common BRAF mutation found in different cancer types results from thymine-to-adenine transversions at nucleotide position 1799, leading to a valine-to-glutamate substitution at residue 600 (V600E). The V600E substitution disrupts the hydrophobic interactions between the activation loop and the ATP binding site that maintain the protein in an inactive conformation, which allows the formation of new interactions that keep the protein in a catalytically competent conformation, resulting in continuous phosphorylation of MEK (164).
Mutations of the BRAF gene are found in 40% to 45% of papillary carcinomas and therefore represent the most common genetic alteration known to occur in thyroid tumors. The spectrum of mutations affecting this gene includes point mutations, small in-frame deletions or insertions, and chromosomal rearrangement. The most common mutation is V600E, which accounts for 98% to 99% of all BRAF mutations found in thyroid cancer (165,166). Other alterations involve K601E point mutation and small in-frame insertions or deletions surrounding codon 600 (167,168,169,170,171), as well as the AKAP9/BRAF rearrangement (172). This rearrangement is a paracentric inversion of chromosome 7q leading to the fusion between the portion of the BRAF gene encoding the protein kinase domain to the AKAP9 gene (172). All point mutations and the rearrangement lead to the activation of BRAF kinase, increase of MEK phosphorylation, and sustained stimulation of the MAPK pathway. The causal role of BRAF mutation in thyroid tumor initiation
has been demonstrated in transgenic and in knock-in mice with thyroid-specific expression of V600E (10,161,173). The animals developed papillary carcinomas with high penetrance and microscopic features similar to those found in human papillary carcinomas. Interestingly, when expressed at physiologic levels in thyroid cells, the mutant BRAF required TSH stimulation to initiate papillary carcinoma development (10). BRAF-induced human and murine PTCs are often locally invasive. Interestingly, expression of oncogenic BRAF in PCCL3 thyroid cells is associated with increased expression of numerous genes involved in extracellular matrix remodeling, including matrix metalloproteinases (174). A role for thrombospondin 1 in invasiveness has also been demonstrated in orthotopic xenografts of a BRAF mutant human thyroid cancer cell line (175).
has been demonstrated in transgenic and in knock-in mice with thyroid-specific expression of V600E (10,161,173). The animals developed papillary carcinomas with high penetrance and microscopic features similar to those found in human papillary carcinomas. Interestingly, when expressed at physiologic levels in thyroid cells, the mutant BRAF required TSH stimulation to initiate papillary carcinoma development (10). BRAF-induced human and murine PTCs are often locally invasive. Interestingly, expression of oncogenic BRAF in PCCL3 thyroid cells is associated with increased expression of numerous genes involved in extracellular matrix remodeling, including matrix metalloproteinases (174). A role for thrombospondin 1 in invasiveness has also been demonstrated in orthotopic xenografts of a BRAF mutant human thyroid cancer cell line (175).
BRAF V600E mutation is typically found in papillary carcinomas with classical histology and the tall cell variant, and is rare in the follicular variant of papillary carcinoma (76,176). BRAF mutations can occur early in development of papillary carcinoma, based on evidence that they are present in microscopic tumors (177). In addition to papillary carcinomas, BRAF V600E mutations are seen in poorly differentiated thyroid carcinomas and anaplastic carcinomas, particularly in those tumors that also contain areas of well-differentiated papillary carcinoma (177,178,179). In these tumors, BRAF mutation is detectable in both well-differentiated and poorly differentiated or anaplastic tumor areas, providing evidence that it occurs early in the tumorigenic process. The properties of mutant BRAF to predispose to tumor dedifferentiation have also been seen in transgenic animals, which develop widely invasive papillary carcinomas with progression to poorly differentiated carcinoma (173). A process of dedifferentiation of BRAF-mutated cancers coincides with profound deregulation of expression of genes involved in cell adhesion and intracellular junction, providing evidence for epithelial-mesenchymal transition in these tumors (180,181).
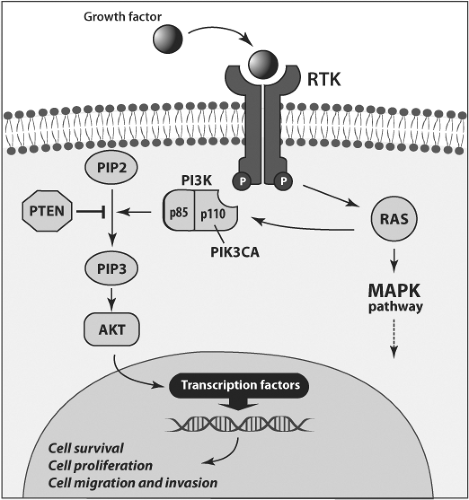 Figure 50B.2 Schematic representation of the PI3K/PTEN/AKT signaling pathway and location of mutations in thyroid tumors. This pathway transmits signals from cell membrane growth factor receptors (RTK), which activate PI3K directly or via RAS. AKT is activated downstream of PI3K and regulates multiple genes that oppose apoptosis and promote cell survival, proliferation, and migration/invasion. PTEN is a dual phosphatase that negatively regulates PI3K signaling and prevents AKT activation. PI3K consists of two subunits, one of which, p110, is coded by the PIK3CA gene. In thyroid tumors, this pathway can be activated by gain-of-function mutations of RAS, PIK3CA, and AKT and loss-of-function mutations of PTEN. PI3K – phosphatidylinositol-3-kinase; PIP2 – phosphatidylinositol-3,4-diphosphate; PIP3 – phosphatidylinositol-3,4,5-triphosphate; MAPK – mitogen-activated protein kinase. |
BRAF V600E has not been found in follicular carcinomas and benign thyroid nodules, and therefore among thyroid lesions this mutation represents a specific marker of papillary carcinoma and related tumor types (176). BRAF V600E mutation may also serve as a prognostic marker for papillary thyroid carcinoma. Its association with more aggressive tumor characteristics, such as extrathyroidal extension, advanced tumor stage at presentation, and lymph node or distant metastases, has been found in most studies involving large series of patients, although some observations have not reported such an association [reviewed in Ref. (182)]. More importantly, BRAF V600E has been found to be an independent predictor of treatment failure and tumor recurrence, even in patients with low stage disease (183,184,185), and as an independent risk factor for tumor-related death (186). The more aggressive behavior of BRAF V600E positive tumors may be due to the propensity of these tumors to dedifferentiate, and due to the fact that this mutation leads to the alteration of expression and/or function of the sodium iodide symporter (NIS) and other genes metabolizing iodide, decreasing the ability of tumors to trap radioiodine and therefore predisposing to the failure of radioiodine treatment (183,187,188,189).
There is practically no overlap between PTC with RET/PTC, BRAF, or RAS mutations, which altogether are found in about 70% of cases (76,165). The lack of concordance for these mutations provides compelling genetic evidence for the requirement of this signaling system for transformation to papillary cancer. As these signaling proteins function along the same pathway in thyroid cells, this represents a paradigm of human tumorigenesis through mutation of three signaling effectors lying in tandem (Fig. 50B.1).
PI3K/AKT Pathway Mutations
The PI3K- protein kinase B (AKT) signaling pathway plays an important role in the regulation of cell survival, proliferation, and migration (190). This pathway can be inappropriately stimulated by activating mutations in the RAS, PIK3CA, and AKT genes or by inactivating mutations in the PTEN tumor suppressor gene (Fig. 50B.2). Induction of this pathway induces thyroid cell proliferation and other effects for which activation of the downstream mammalian target of rapamycin (mTOR) protein appears particularly critical (191).
Mutations in the effectors of this signaling pathway occur in 5% to 10% of well-differentiated follicular carcinomas and are rarely found in papillary cancers. These mutations are significantly more common in anaplastic and poorly differentiated carcinomas, where 10% to 20% of tumors harbor PIK3CA mutations and 5% to 10% AKT1 mutations (189,192,193,194,195). The majority of PIK3CA mutations are located at exon 20, the kinase domain, and exon 9, the helical domain, and lead to the activation of this pathway as demonstrated by the increased phosphorylation of the AKT protein (192). In addition to point mutations, an increase in PIK3CA gene copy number is found in ∼40% of anaplastic carcinomas (193,196). However, whether these copy number gains are functionally relevant remains unclear, as it has not been shown that these are the result of focal chromosomal amplification events, or that these particular cancers are dependent on the activity of the pathway for viability.
PI3K pathway gene mutations are most likely a late event in thyroid carcinogenesis. They occur with higher prevalence in advanced and anaplastic carcinomas as compared to well-differentiated carcinomas. Moreover, in contrast to BRAF and RAS mutations which almost never coexist in the same tumor, PIK3CA and AKT1 mutations are frequently seen in anaplastic carcinomas coexisting with BRAF or RAS mutations (192,193). Moreover, PIK3CA and particularly AKT1 mutations have been reported in metastatic thyroid cancers from primary tumors that lack these mutations (185,189). There is also supportive evidence from mouse models demonstrating the relevance of AKT activation for development of metastases (126,197).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


