Fig. 3.1
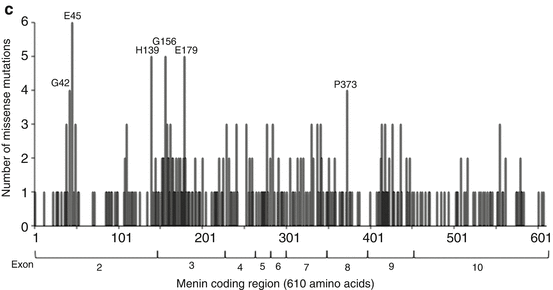
MEN1 gene and missense mutations. (a) Genomic structure of the MEN1 gene. The 10 exons of the gene are shown as boxes that are sized with the indicated 100-bp scale. The broken lines in-between the boxes are introns, not drawn to scale. Striped parts indicate untranslated regions that are not present in the menin coding region, marked with the start codon ATG in exon 2 and stop codon TGA in exon 10. The two nuclear localization signals (NLS) at amino acids 479–497 and 588–608 are marked in exon 10. (b) Germline and somatic missense mutations in the MEN1 gene. The menin coding region consists of 610 amino acids (aa) that starts in exon 2 and ends in exon 10. The extent of the coding region in each exon is as follows: ex2(aa1-148), ex3(aa149-218), ex4(aa219-261), ex5(aa262-275), ex6(aa276-304), ex7(aa305-350), ex8(aa351-395), ex9(aa396-450), and ex10(aa451-610). Missense mutations in each exon are shown. The germline and somatic missense mutations have been assembled from References 35 and 36 and from the COSMIC database. There are 340 different missense mutations affecting 239 amino acids. The amino acids are numbered as per menin transcript variant-2 that has 610 amino acids. Note that menin transcript variant-1 has 615 amino acids. Readers should be careful about the interpretation of MEN1 mutation nomenclature whether the nucleotides and amino acids are numbered as per transcript variant-1 or variant-2, particularly for mutations located after exon 2 (see explanation in the text). (c) Location of germline and somatic missense mutations in menin. Menin missense mutations at each amino acid (from Fig. 3.1b) are plotted to show the number of unique mutations in each amino acid. Exons are marked below the mutation plot. Missense mutations are distributed over the entire 610 amino acid coding region indicating that all parts of menin are essential. There are six amino acids affected by more than three different mutations (amino acids G42, E45, H139, G156, E179, and P373)
3.3.2 MEN1 Germline Mutations
Germline heterozygous inactivating mutations in MEN1 are observed in 70–90 % of familial MEN1 patients that predisposes to tumor development [29–32]. The frequency of detecting a germline MEN1 mutation is much lower in cases without a family history of MEN1 and in similar sporadic MEN1 cases who present with hyperparathyroidism and pituitary tumor but no PNETs [33, 34]. MEN1-related tumors show LOH at chromosome 11q13 from loss of the region corresponding to the normal non-mutated MEN1 allele [29, 30]. Thus MEN1 acts as a tumor suppressor gene, and its biallelic loss/inactivation leads to tumorigenesis. More than 1000 mutations have been reported in the MEN1 gene [35]. Two studies have reviewed the published germline MEN1 mutations from 1997 to 2007 and 2007 to 2015, and they found a total of 576 unique mutations [35, 36]. A vast majority (65–70 %) of the mutations predict C-terminal truncation of menin or deletion of internal exons encoding menin (nonsense, frameshift, and splicing). Other mutations observed are missense (25 %), in-frame deletion/insertion (5 %), and gross deletions (2 %) [35–37]. The expression of some menin missense mutant proteins has been shown to be unstable and degraded by the proteasome [38]. There is a need to develop assays to study the adverse physiological effect of rare MEN1 mutations, especially for mutations that do not delete a large region of menin, such as missense and single amino acid in-frame del/ins. Such assays may help to determine whether mutations are pathologic or benign.
3.3.3 Genetic Screening of MEN1 Patients
Genetic testing for germline MEN1 mutations helps to confirm the clinical diagnosis, to identify at-risk individuals in a family, and to exclude noncarriers [3, 5]. Tumors and biochemical features of MEN1 are typically established at age 40–50 years; however, a few children before the age of 10 years have been reported with MEN1-related tumors [39–41]. This observation justifies the recommendation of genetic testing at the earliest opportunity [3, 5]. Patients with features of MEN1 but who do not test positive for germline MEN1 mutations by the standard genetic testing methods (that screen the menin coding region and gross deletions) may show germline heterozygous mutations in the CDKN1B/p27 gene or probably in similar cyclin-dependent kinase inhibitor (CDKI) genes p15, p18, or p21 [42–44]. The MEN1-like syndrome with p27 mutations has been named MEN4 [37, 45]. Approximately 1–2 % of MEN1-like cases have been reported to possess germline heterozygous p27 mutations, and less than 1 % with probable p15, p18, or p21 mutations [5, 46]. The CDKI genes of the INK4 family (p15, p16, p18, and p19) and the Cip/Kip family (p21, p27, and p57) are cell cycle inhibitors for specific cyclin-CDK complexes [47]. No genotype–phenotype correlation has been observed for p27 mutation with MEN1 or MEN1-like features.
3.3.4 Somatic Mutations in Sporadic Tumors
MEN1-related tumors can also occur sporadically. Some of these non-inherited tumors show somatic inactivating mutations in MEN1 with or without 11q13 LOH. The prevalence of MEN1 mutations in sporadic tumors is glucagonoma (60 %), VIPoma (57 %), nonfunctioning pancreatic tumor (44 %), gastrinoma (38 %), bronchial carcinoid (35 %), parathyroid adenoma (35 %), lipoma (28 %) insulinoma (2–19 %), angiofibroma (10 %), anterior pituitary tumor (3.5 %), and adrenocortical tumor (2 %) [37, 48–54]. Exome sequencing of sporadic PNETs has shown that 43 % of nonfunctioning PNETs have mutations in chromatin remodeling factors DAXX and ATRX [50], and 13–33 % of functioning PNETs (insulinomas) have a recurrent somatic heterozygous missense mutation T372R in the transcription factor YY1 [54–56].
3.3.5 MEN1 Mutation Analysis
Mutations in the MEN1 gene (germline or somatic) are dispersed over the entire coding region and with no obvious genotype–phenotype correlation with an individual sporadic or MEN1-related tumor, or MEN1-related tumor spectrum, or with specific clinical/biochemical features [4, 35, 36]. To date 340 unique missense mutations have been reported that affect 239 amino acids of menin ([35, 36] + additional somatic from the COSMIC database [57]). The location of all the missense mutations (Fig. 3.1b) illustrates how the mutations hit just about every region of menin (Fig. 3.1c) implying that all regions of menin are essential.
3.4 Structure and Function of Menin
3.4.1 Menin Subcellular Localization and Structure
Menin is a ubiquitously expressed 67-kDa protein that is predominantly localized in the nucleus due to the presence of two nuclear localization signals (NLS), NLS1 (amino acid residues 479–497) and NLS2 (amino acid residues 588–608) [58]; a third accessory NLS, NLSa (amino acid residues 546–572), has also been reported [59]. Menin is highly conserved in animal species but with no known homologs in yeast and nematodes. The crystal structure of Nematostella menin (the starlet sea anemone) and human menin has been deciphered, and they are very similar [60, 61]. The structure resembles a curved left hand, with a deep pocket formed by the thumb and the palm [60, 61]. Menin is an α-helical protein with 4 domains: a long β-hairpin N-terminal domain, a transglutaminase-like domain that forms the thumb, a helical palm domain that contains three tetratricopeptide motifs, followed by a C-terminal fingers domain [60, 61]. The deep cavity in the palm may act as a binding site for interacting proteins [60, 61]. Both structures lack some parts of menin that have been predicted as disordered regions – in Nematostella menin, an unstructured loop (amino acid residues 426–442) and the C-terminus (amino acid residues 487–539), and in human menin, an internal unstructured loop (amino acid residues 460–519) [60, 61]. Therefore, the crystal structure of the entire menin protein in its free form or together with an interacting partner remains to be determined.
3.4.2 Menin-Interacting Proteins
Menin does not show homology to any known proteins and it does not possess any enzymatic activity. Therefore to elucidate the function of menin, interacting proteins have been sought, and approximately 40 different proteins have been shown to partner with menin – transcription factors, chromatin modification factors, transcription initiation and elongation factors, cytoskeletal proteins associated with cell division/adhesion/motility, DNA repair proteins, an mRNA biogenesis factor, and signaling mediators in the cytoplasm (Fig. 3.2) [37, 62–64]. These interactions predict that menin may participate in a variety of functions in different cell types. A majority of the interactions are with proteins involved in transcriptional regulation; however, menin by itself does not possess a DNA-binding domain to directly regulate transcription [60, 61]. Protein and DNA interaction studies have revealed some menin target genes (cell cycle inhibitors p18 and p27, the long noncoding RNA MEG3, IGFBP2, and the HOX genes) that are regulated by menin from interactions with the mixed lineage leukemia (MLL) protein in a protein complex that is responsible for a specific chromatin mark of gene activation, histone H3 lysine 4 trimethylation (H3K4me3) [65–73]. Loss of menin results in loss of H3K4me3 at specific genes and, at the same genes, gain of H3K27me3 that is an epigenetic mark of gene repression [71]. Phosphorylation at six different amino acid residues of menin has been reported – Ser394, Thr397, Thr399, Ser487, Ser543, and Ser583 – constitutively (Ser543) or in response to DNA damage (Ser394 and Ser487) or upon loss of phosphorylation at Ser394 (Thr397 and Thr399) [74]. Menin is reported to undergo another posttranslational modification, SUMOylation at Lys591 [75]. The impact of phosphorylation or SUMOylation on the crystal structure of menin or in protein–protein interactions is not known. Most of the protein interaction studies have been conducted in cell lines unrelated to NETs or other tumors of MEN1 [76]. In order to confirm the MEN1-related relevance of the functions of menin through its interacting proteins, there is a need to develop cell lines from tissues (normal and tumor) affected in MEN1. Such studies may help to define the biological functions of menin that are critical to prevent the development of MEN1-related tumors.

Fig. 3.2
Menin interacting proteins. More than 40 different protein partners of menin are shown. The proteins are divided into functional categories. Majority of the interactions are observed in the nucleus with proteins involved in transcriptional regulation. Menin binding regions of three proteins have been shown in co-crystal structures with menin (proteins marked in bold with an asterisk). Similar analysis of the other menin interacting proteins has not been performed
3.5 Molecular Genetics of MEN1-Related NETs in Mouse Models
3.5.1 Germline Men1 Knockout Mice
Germline homozygous loss of the Men1 gene (Men1−/−) in mice results in death in utero between embryonic days 10.5 and 14.5, but germline heterozygous loss of the Men1 gene (Men1+/−) does not cause embryonic lethality [77–81]. After 9–16 months, Men1(+/−) mice develop MEN1-related endocrine tumors: parathyroid tumors, pancreatic islet tumors (mainly insulinoma), and anterior pituitary tumors (mainly prolactinoma in female) [77, 79–81]. Gastrinomas are seen in only one of the four different mouse models of MEN1 [79]. Foregut carcinoids are not seen in the mouse models of MEN1. Other tumors observed in the Men1(+/−) mouse model of MEN1 are adrenal cortical tumors, gonadal tumors (Leydig cells in male or ovarian stroma in female), and bilateral pheochromocytoma [82, 83]. Similar to MEN1-related human tumors, the tumors in Men1(+/−) mice develop after loss of the wild-type Men1 allele (LOH). Given that menin may regulate the expression of cell cycle genes, mouse models with loss of tumor suppressor genes known to regulate the cell cycle (Rb or p53) or cell cycle regulator genes (Cdk2, Cdk4, p18, or p27) have been generated in the Men1(+/−) background to study the impact on the development of MEN1-related NETs, PNETs in particular (Table 3.1). Combined loss of Men1(+/−) with Rb or p53 did not show any significant effect [84–86]. Combined loss of Men1(+/−) with p18 but not p27 accelerated the rate of insulinoma formation and with an increased tumor incidence [87]. Mice with combined loss of Men1(+/−) with Cdk4 but not Cdk2 did not develop insulinoma [88]. These mouse models show that p18 inactivation and Cdk4 activation may be critical for PNET formation upon menin loss.
Table 3.1
PNETs (insulinoma) in mouse models of Men1 loss upon combined genetic manipulation with candidate genes
Genotype | Phenotype | Ref. |
|---|---|---|
Men1(+/−) | Parathyroid hyperplasia, anterior pituitary tumor, and pancreatic islets tumors (insulinoma) at ages 12–18 months Tumors with Men1 LOH | |
p18(−/−) | Islet tumor phenotype similar to Men1(+/−) | [87] |
p18(−/−); Men1(+/−) | Develop islet tumors at an accelerated rate and with an increased incidence Tumors without Men1 LOH | [87] |
p27(−/−) | Do not develop islet hyperplasia | [87] |
p27(−/−); Men1(+/−) | Islet tumorigenesis comparable to Men1(+/−) | [87] |
Cdk2(−/−) | No developmental or functional defects in neuroendocrine tissues | [88] |
Cdk2(−/−); Men1(+/−) | Islet tumorigenesis comparable to Men1(+/−) Tumors with Men1 LOH | [88] |
Cdk4(−/−) | Hypoplasia of the anterior pituitary and pancreatic islets during postnatal periods | [88] |
Cdk4(−/−); Men1(+/−) | Do not develop any tumors; islets and pituitaries remained hypoplastic No LOH for Men1 | [88] |
Rb(+/−) | Hyperplasia of pancreatic islets | |
Rb(+/−); Men1(+/−) | Islet tumorigenesis comparable to Men1(+/−) Tumors with Men1 LOH | |
p53(+/−) | Do not develop islet tumors | [86] |
p53(+/−); Men1(+/−) | Islet tumorigenesis comparable to Men1(+/−) | [86] |
Men1(f/f); RIP-Cre | Pancreatic islet tumors (insulinoma) at ages 6–8 months and anterior pituitary tumors | |
K-RAS(G12D) | K-RAS(G12D) inhibited β-cell proliferation (P5 neonates) | [101] |
K-RAS(G12D); Men1(+/f); RIP-Cre | K-RAS(G12D) enhanced, rather than inhibited β-cell proliferation (P5 neonates) | [101] |
Ctnnb1(f/f); RIP-Cre | No significant effect of β-catenin loss on mature islets | [102] |
Ctnnb1(f/f); Men1(f/f); RIP-Cre | Loss of β-catenin suppressed islet growth and tumorigenesis | [102] |
Rbp2(f/f); RIP-Cre | Rbp2 loss did not grossly affect islet histology or function | [103] |
Rbp2(f/f); Men1(f/f); RIP-Cre | Decreased islet tumor formation and prolonged survival | [103] |
Inhβ(−/−) | No significant effect of ActivinB loss on mature islets | [108] |
Inhβ(−/−); Men1(f/f); RIP-Cre | Prolonged survival after 10 months of age | [104] |
Men1(f/f); PDX1-Cre | Selective development of a single islet tumor (insulinoma) | [90] |
Arc(−/−) | No basal phenotype from Arc loss | [109] |
Arc(−/−); Men1(f/f); PDX1-Cre | Did not significantly alter tumor load | [109] |
3.5.2 Tissue-Specific Men1 Knockout Mice
Mice with tissue-specific homozygous loss of Men1 in the parathyroids, pancreas, or pancreatic islet β-cells are viable and develop tumors in the specific tissue at an earlier age than in the Men1(+/−) mice [89–93]. The exception is a mouse model with liver-specific homozygous Men1 knockout that lacks tumors in the liver, a tissue not associated with tumors in MEN1 patients, underscoring tissue-specific actions of menin in MEN1-related target tissues [94]. The tissue-specific action of menin as a tumor suppressor in the pancreatic β-cells is highlighted by the observation that homozygous loss of Men1 in β-cells (Men1(f/f); RIP-Cre) or in the whole pancreas (Men1(f/f); PDX1-Cre), both show only β-cell tumors (insulinomas) [90–93]. Another interesting observation in mouse models is that mice with pancreatic α-cell-specific loss of Men1 develop insulinomas rather than the expected glucagonomas due to possible transdifferentiation of α-cells into β-cells, or perhaps paracrine signals that induce β-cell proliferation [95, 96]. The molecular mechanisms underlying β-cell-specific tumorigenesis from menin loss remain to be determined. Perhaps menin has tissue-specific interactions or regulates tissue-specific factors as indicated in a few studies that have implicated β-cell-specific differentiation factors MafA or Hlxb9/Mnx1 [97–100]. Effect of menin loss together with target genes associated with β-cell proliferation and function has been studied in mouse models (Table 3.1). Combined genetic manipulation of candidate genes in mice with β-cell-specific Men1 knockout (Men1(f/f); RIP-Cre) shows that expression of activated K-RAS(G12D) enhances rather than inhibits β-cell proliferation, β-catenin loss can suppress islet growth and tumorigenesis, histone demethylase Rbp2 loss decreases islet tumor formation and prolongs survival, and ActivinB loss prolongs survival after 10 months of age [101–104]. Further studies of these mouse models may help to understand β-cell-specific disease-associated pathways and to develop potential therapies for MEN1-related PNETs.
3.5.3 Men1 Knockout Mice for Preclinical Studies
Mouse models of Men1 loss have served as preclinical models to study the potential of various treatment options in PNETs (insulinoma) and pituitary tumors (prolactinoma) – an angiogenesis inhibitor (anti-VEGF-A monoclonal antibody, mAb G6-31), a small molecular tyrosine kinase inhibitor of all VEGF receptors (sunitinib), a somatostatin analog (pasireotide/SOM230), and menin replacement therapy [105–107]. Similar preclinical investigations may be possible from the results described above about studies in mouse models with combined loss of Men1 and genetic manipulation of candidate genes such as inhibitors of CDK, β-catenin, ActivinB, or histone demethylation.
3.6 Conclusions
Patients with the MEN1 syndrome can undergo germline genetic testing for the MEN1 gene that can help to confirm the clinical diagnosis, to identify at-risk individuals within families for early monitoring of tumors, and to exclude individuals who will not develop tumors. However, 10–30 % of cases with clinical MEN1 features lack germline MEN1 mutations. To identify the causative gene/s in these MEN1 mutation-negative cases, studies can be conducted by using whole genome/exome sequencing approaches to analyze germline DNA of patients and family members with different combinations of MEN1-related tumors. Molecular genetic studies in cell lines and mouse models have revealed candidate genetic interactions, many protein partners, and menin-dependent target genes. There is a need for integrated analysis of data from these studies that will help to define the exact role/s of menin in normal physiology and the molecular events after menin loss that lead to tumor formation in specific tissues. Such a comprehensive understanding of the process of tumorigenesis in MEN1-related NETs in man and mouse can provide considerable support to translational efforts for developing antitumor therapeutic options in human patients. In contrast to NETs in patients without MEN1, MEN1-related NETs are frequently multiple and with an earlier age of onset and with a potential for malignancy; therefore, screening and treatment options specific for MEN1-related NETs have to account for these differences.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases (Project number: 1ZIADK075035-07).
References
2.
Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129:484–94.PubMed
3.
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–71.PubMed
Related posts:
 Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
 Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
 Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
Radionuclide Imaging of Pancreatic Neuroendocrine Tumours
 Nanoparticles for Radionuclide Imaging and Therapy: Principles
Nanoparticles for Radionuclide Imaging and Therapy: Principles
 Radionuclide Imaging of Medullary Thyroid Carcinoma
Radionuclide Imaging of Medullary Thyroid Carcinoma
 Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


