Figure 26-1 Kaplan-Meier analysis of overall survival in 2628 children with newly diagnosed acute lymphocytic leukemia (ALL). The patients participated in 15 consecutive studies conducted at St. Jude Children’s Research Hospital from 1962 to 2005. The 5-year overall survival estimates (± SE) are shown, except for Study 15, for which preliminary results at 4 years are provided. (From Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166-178, with permission.)
Abnormalities of Chromosome Number (Ploidy)
Ploidy can be assessed either by chromosome number or by flow cytometry using the DNA index (DI), the ratio of fluorescence in leukemic blasts compared to normal cells. Normal diploid cells have 46 chromosomes and a DI of 1.0, hyperdiploid cells have higher values, and hypodiploid cells lower. Hyperdiploidy is further classified as “low” and “high” (greater than 50 chromosomes).
Hypodiploid cases constitute approximately 6% of pediatric and 2% to 8% of adult ALL. 5 Those with fewer than 45 chromosomes have significantly worse outcome, with the worst outcome occurring in near-haploid cases (24 to 28 chromosomes). 5 The adverse prognostic impact in adults is somewhat weaker. “Pseudodiploid” cases, with normal chromosome number but structural abnormalities, also do relatively poorly. Rare cases with near triploidy or near tetraploidy (more than 80 chromosomes) have traditionally been associated with poor outcome. However, more recent reports analyzed from studies using modern therapies refute this claim, showing that these lesions should be classified prognostically as neutral 6 or even favorable. 7
Hyperdiploidy occurs in about 35% of pediatric and 25% of adult ALL cases. 8 In children, ALL with more than 50 chromosomes, or simply the simultaneous trisomies of 4 and 10 (and less importantly 17), is an independent positive prognostic indicator. In adults, the prognostic implications are less clear. Although the biologic basis of hyperdiploidy is poorly defined, it often co-occurs with other favorable risk factors as well as Ras and FLT3 mutations and FHIT hypermethylation.
Genetic Abnormalities in ALL
B-ALL
Abnormalities of Chromosome Structure
(ETV6-RUNX1), t(12;21)(p13;q22)
The ETV6-RUNX1 (TEL-AML1) fusion protein formed by the t(12;21)(p13;q22) translocation is the most frequent abnormality in children (25%), whereas it is much rarer in adults (2%). ETV6 (ETS variant gene 6) is also known as TEL (translocation-ETS-leukemia). RUNX1 (runt-related transcription factor 1) is also known as AML1 or CBFA2 (core binding factor A2). In nearly all cases the translocation is cryptic, involving a region too small to be detected by karyotype.
ETV6 encodes a widely expressed nuclear protein belonging to the Ets family of transcription factors, which are involved in diverse developmental processes including the establishment of embryonic and adult hematopoiesis. A helix-loop-helix (HLH) region known as the ETS domain allows DNA binding for transcriptional regulation, and an HLH region known as the pointed domain appears to facilitate self-association (Figure 26-2 , A). RUNX1 is a transcription factor with highly restricted expression in hematopoietic cells and developing ganglions but that is required for transcription of several hematopoietic-specific genes, and may organize the factor complex necessary for lineage-specific transcription.
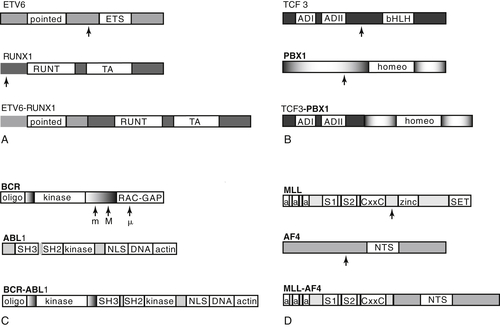
Figure 26-2 Schematic of key domains of the genes involved in several principal translocations in acute lymphocytic leukemia (ALL) and the translocation products. Arrows indicate common breakpoints. Note that loss of the domain conferring sequence specificity to transcription factor binding occurs in ETV6-RUNX1 (ETS domain of TEL) and TCF3-PBX1 (bHLH domain of E2A). Gene regions are not drawn to scale. (A) ETV6-RUNX1. TA, Transactivation domain. (B) TCF3-PBX1. ADI and ADII, Activation domains I and II; bHLH, basic helix-loop-helix, sequence-specific DNA-binding domain; homeo, homeobox domain. (C) BCR-ABL. All three breakpoint regions are indicated (M, major, for p210 protein; m, minor, for P190 protein; μ, associated with P230 protein). Only the P190 fusion product is illustrated. oligo, Oligomerization domain; kinase, serine-threonine kinase domain; RAC-GAP, RAS-like GTPase; SH3, SH2, and kinase, SRC homology domains; kinase, tyrosine kinase domain; NLS, nuclear localization domains; DNA, DNA-binding site; actin, G and F actin binding site. (D) Mixed lineage leukemia. a, AT-hook; CxxC, cysteine-rich motif homologous to DNA methyltransferase; S1 and S2, subnuclear localization domains; NTS, nuclear targeting sequence; zinc, zinc fingers.
The ETV6-RUNX1 fusion protein is widely expressed because of the ETV6 promoter and converts RUNX1 from a transcriptional activator to a repressor. The exact mechanism of repression is unclear, as is the manner in which this mediates leukemogenesis. ETV6-RUNX1 overexpression causes leukemia in only a minority of mouse models, with low penetrance and prolonged latency, suggesting that additional events are crucial for full transformation. A frequent secondary event in ETV6-RUNX1+ ALL is loss of heterozygosity (LOH), deletion, or otherwise downregulated expression of the remaining normal copy of ETV6, suggesting a potential role for ETV6 as a tumor suppressor.
ETV6-RUNX1 positivity tends to occur in children 1 to 10 years of age, and nearly exclusively in CD10+ B-precursor ALL. In the past, cases characterized by ETV6-RUNX1 + ALL had a hallmark tendency to relapse late, with excellent chemosensitivity and salvage rate. On modern treatment regimens ETV6-RUNX1 relapsed disease is extremely rare and, although survival for all subtypes of ALL in children with contemporary chemotherapy strategies has improved, ETV6-RUNX1 has retained positive prognostic significance. 9 When relapse does occur, evidence suggests that it may represent evolution of a new leukemic clone from the preleukemic ETV6-RUNX1 + cell of origin.
TCF3-PBX1, t(1;19)(q23;p13)
The TCF3-PBX1 fusion protein, associated with the t(1;19)(q23;p13) translocation, is the second most common translocation in pediatric ALL, occurring in approximately 6% of all pre-B ALL. 10 It is a rare (3%) and adverse feature in adults. The fusion protein combines the two activation domains of the basic helix-loop-helix (bHLH) transcription factor TCF3 (previously E2A) on chromosome 19 with the homeobox (HOX) gene PBX1 (for pre-B cell homeobox 1) on chromosome 1, resulting in a strong transcriptional activator effect on PBX1 (see Figure 26-2, B). TCF3 is a transcriptional activator critical in lymphocyte development, as well as widely expressed and influential in diverse cellular processes. PBX1 belongs to the TALE (three amino acid loop extension) class of atypical homeodomain proteins. The homeodomain mediates both DNA-binding and HOX gene interaction.
The TCF3-PBX1 chimeric transcription factor strongly activates a subset of HOX genes normally regulated by PBX1. The basis for its transforming ability may be reduction of wild-type TCF3 levels; aberrant activation of PBX1 targets in pre-B cells; or activation of targets not normally regulated by PBX1 that are affected by the TCF3-PBX1 fusion protein. 11 Fusion protein overexpression in mouse models causes a variety of leukemias, although not B-lineage ALL, suggesting potent non–lineage-specific transforming activity. Unlike other ALL translocations, t(1;19)+ ALL does not show evidence of in utero origin.
TCF3-PBX1 positivity often coincides with other high-risk factors. Early studies indicated an independent adverse prognostic impact, but on modern intensive pediatric regimens, survival is equivalent. 10 The t(1;19) occurs most often as an unbalanced translocation. Cases with a balanced translocation do more poorly in some studies but not others.
TCF3-HLF, t(17;19)(q23;p13)
The t(17;19) translocation is a much rarer event, occurring in approximately 1% of pediatric ALL and rarely in adults. 12 The HLF (for hepatic leukemia factor) fusion partner is a transcription factor not normally expressed in hematopoietic cells. Potential oncogenic effects of the fusion protein include repression of normal TCF3 function, altered transcriptional activity of HLF, and promotion of lymphoblast survival, possibly as the result of antiapoptotic effects. TCF3-HLF ALL tends to occur in adolescents and is frequently associated with hypercalcemia, disseminated intravascular coagulopathy (DIC), a cIgM-negative, low CD10 positivity pro-B cell immunophenotype, and a poor prognosis despite intensive chemotherapy.
BCR-ABL1, t(9;22)(q34;q11)
The t(9;22) translocation was the first recurrent chromosomal abnormality identified in human cancer, in association with chronic myelocytic leukemia (CML). This translocation, known as the Philadelphia chromosome (Ph), is necessary and sufficient in transformation to the preleukemic, myeloproliferative neoplasm CML. BCR-ABL1 is also the most common translocation noted in adult ALL, with a prevalence of 25%, where it is known to be leukemogenic but not in itself sufficient to cause disease. The occurrence of the Ph in ALL increases with age and is therefore seen only in about 3% of childhood ALL cases. 13 Philadelphia-positive ALL is primarily a CD10+ precursor B ALL with frequent coexpression of myeloid markers. Patients with Ph+ ALL tend to be older, present with higher leukocyte and peripheral blast counts, have central nervous system (CNS) involvement, and historically have exhibited lower induction remission rates, shorter remission durations, and very poor overall survival (OS). Monosomy 7 and/or loss of 9p are secondary aberrations that may be associated with worse outcomes.
The Philadelphia chromosome is formed by in-frame fusion of the 5′ portion of BCR (for breakpoint cluster region) on chromosome 22 to the 3′ portion of the tyrosine kinase C-ABL1 on chromosome 9, a proto-oncogene that is part of the RAS signaling pathway (see Figure 26-2, C). The fusion protein upregulates ABL1 tyrosine kinase activity. Two main fusion proteins occur, which differ in BCR breakpoint. Breaks within the 5.8-kb major breakpoint cluster region (M-BCR), occurring in CML and 25% of adult Ph+ ALL, form a 210-kDa protein known as p210. In the remainder of adult ALL and the majority of pediatric ALL, the breakpoint occurs further upstream, in the minor breakpoint cluster region (m-BCR), forming a 185- to 190-kDa protein usually known as p190. An additional breakpoint generates a 230-kDa protein associated with a rare CML variant with neutrophilia and occasionally with classic CML. All three transcripts can be detected at very low levels using sensitive PCR techniques. It has been suggested that the p190 protein arises de novo, whereas the p210 protein may represent the blast crisis of a previously unrecognized CML. Other features that may distinguish cases that originated as CML include basophilia, marked splenomegaly, and persistence of the BCR-ABL1 fusion protein in hematopoietic precursor cells of all lineages following remission.
Treatment of CML and Ph+ ALL was revolutionized by the development of imatinib mesylate, also known as STI-571 or Gleevec, introduced in 2001. 14 Imatinib, a selective tyrosine kinase inhibitor, was the first molecularly targeted therapy to attain large-scale clinical success, fulfilling the goals of antitumor selectivity and low systemic toxicity. Despite its success, however, it has not been effective as a single agent because of the rapid development of resistance. Nevertheless, in ALL, when combined with standard cytotoxic chemotherapy, imatinib has dramatically improved patient outcomes. In adults, imatinib plus chemotherapy alone or as a bridge to transplant has improved 4-year OS from roughly 15% to between 38% and 54%. 15 In children, outcomes with the addition of imatinib have been even more striking. Children’s Oncology Group (COG) study AALL0031 demonstrated that addition of imatinib to high-intensity chemotherapy improved 3-year EFS to 80% compared to historical controls of 35%. 13 Outcomes in patients treated with chemotherapy plus imatinib were similar to those in patients receiving sibling donor bone marrow transplant (BMT), suggesting that BMT in first remission may no longer be the treatment of choice for Ph+ ALL in children. 13 New developments in the treatment of Ph+ ALL include more potent second-generation tyrosine kinase inhibitors; dual SRC and BCR-ABL1 kinase inhibitors; and combination therapy with a farnesyl transferase or a PI3 kinase inhibitor.
MLL, 11q23 Rearrangements
MLL gene rearrangements (MLL-r) occur in 8% of pediatric ALL and 10% of adult ALL and constitute the most frequent abnormality in infant ALL, occurring in 60% to 70% of cases. 16 MLL-r is also associated with AML, particularly secondary malignancies following anthracyclines and epipodophyllotoxins. MLL-r leukemias are unusual in two respects: the N terminus of MLL forms a fusion protein with the C termini of more than 70 different partners, including itself; 17 and MLL-r are found in both ALL and AML, whereas most other translocations are lineage specific. Indeed, MLL takes its name, “mixed lineage leukemia,” from this distinguishing feature (it is also known as HRX or HTRX for homology to trithorax in Drosophila, and ALL1 for involvement in ALL). MLL-r ALL has a unique gene expression pattern suggestive of arrest at an earlier hematopoietic progenitor stage. The MLL-AF4 fusion protein formed by t(4;11) is the most common MLL translocation in ALL, making up 70% of cases. The MLL-ENL fusion formed by t(11;19) comprises another 13%.
Infant ALL with MLL-r tends to be associated with age less than 6 months, massive tumor burden, organomegaly, frequent CNS involvement, coexpression of myeloid antigens, and CD10-negative pro-B immunophenotype. MLL-r is a very poor prognostic feature in infant ALL, and a poor prognostic feature in children more than 1 year of age. One surprising exception is MLL-ENL, associated with good prognosis in both T ALL and patients age 1 to 9 years. 18
MLL is a large protein consisting of multiple motifs, including AT-hook DNA-binding domains, transcriptional activation and repression zinc finger domains, and a highly conserved SET domain that regulates homeotic (Hox) promoters. Several motifs are homologous to the Drosophila trithorax protein, which maintains appropriate expression of the Hox genes controlling segment determination. In normal hematopoiesis, MLL is required to generate stem-cell progenitors and to establish both lymphoid and myeloid lineages. In general, the many MLL fusion partners fall in the broad categories of signaling molecules and nuclear transcription factors.
Because Hox genes can induce leukemia, it is thought that the MLL-r protein mediates leukemogenesis through disruption of normal Hox expression patterns. The contributions of MLL’s many fusion partners to leukemic transformation remain unclear, as they do not all share structural or functional similarities (see Figure 26-2, D). One common function may be converting MLL to a constitutive transcriptional effector. In addition, there is a growing body of evidence that MLL-r leukemias are driven primarily by epigenetic dysregulation, including aberrations in DNA and histone methylation and histone acetylation. 19
Both infant and treatment-related leukemias show nonrandom clustering of MLL breakpoints within exons distinct from the breakpoints in de novo MLL-r leukemias, suggesting a shared mechanism of leukemogenesis. One hypothesis is that infant ALL arises from in utero exposure to topoisomerase II inhibitors, analogous to the initiating insult in therapy-related MLL-r leukemias. Epidemiologic studies of the association between infant leukemia and in utero exposure to topoisomerase II inhibitors have identified a modest association between dietary intake and MLL-r AML, and a stronger association between certain medications and insecticides and MLL-r ALL. 20
Event-free survival in infant ALL is generally poor, ranging from 20% to 40%, with t(4;11) having a particularly dismal prognosis. Induction rates are generally comparable to other types of ALL, but early relapse is frequent, usually within a year of diagnosis. MLL-r ALL shows relative resistance in vitro to glucocorticoids and L-asparaginase, and sensitivity to cladribine and Ara-C. High-dose Ara-C has been incorporated into infant ALL regimens with modest success. The receptor tyrosine kinase FLT3 is highly expressed in MLL-r ALL 21 and the use of FLT3 inhibitors and other novel molecularly targeted therapies is under investigation.
Intrachromosomal Amplification of Chromosome 21 (iAMP21)
Recently, a novel recurrent chromosomal abnormality, the intrachromosomal amplification of chromosome 21 (iAMP21), has been identified in a rare (1.5% to 2% of ALL) and very poor prognostic subgroup of patients with ALL. In childhood ALL, iAMP21 patients tend to be older, with lower initial white blood cell and platelet counts. Clinical outcomes are poor, with relapse rates ranging from 38% to 61% depending on the treatment regimen. 22 Most known abnormalities take place in a 5.1-Mb common region of amplification (CRA) on the long arm of chromosome 21 that includes RUNX1, miR-802, and genes mapping to the Down syndrome critical region (DSCR). ALL with iAMP21 consistently has multiple copies of RUNX1 identified in array-based comparative genomic hybridization and FISH assays (Figure 26-3 , A). However, gene expression assays fail to show either overexpression of RUNX1 RNA or a unique iAMP21 signature as compared to other ALL samples. Although the basis for the increased risk of relapse in this subgroup remains unclear, iAMP21 is now used to stratify patients to receive intensified chemotherapy on several current ALL treatment protocols. 23
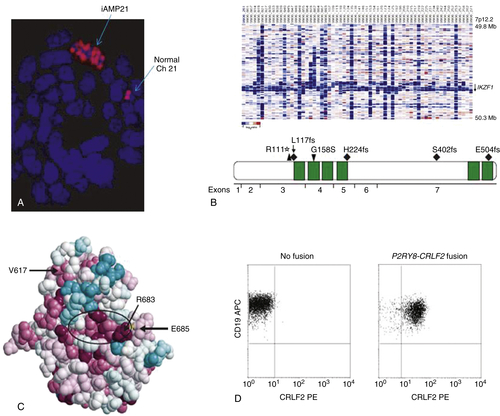
Figure 26-3 Recently identified chromosomal aberrations in ALL (A) iAMP21: Metaphase FISH demonstrates multiple RUNX1 signals (red) on the chromosome 21 with the iAMP21 aberration (top), whereas a single signal is noted on each normal chromosome 21 (right). (Modified from Harrison CJ. Cytogenetics of pediatric and adolescent acute lymphoblastic leukemia. Br J Haematol. 2009;144:147-156, with permission.) (B) IKZF1 deletions and mutations: DNA copy number heatmap (top) demonstrating deletions (blue) at the IKZF1 locus in a cohort of ALL cases, and primary protein structure of IKAROS (bottom) showing the location of the six zinc fingers (green) and missense (downward-pointing arrowhead), frameshift (diamonds), and nonsense (upward-pointing arrowhead) mutations observed in six ALL cases. (From Mullighan C, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470-480, with permission.) (C) JAK2 mutations: Amino acid structural model showing that R683, the amino acid most frequently affected by point mutations in ALL, is situated on a rim of highly conserved (red), exposed amino acids that make up an apparent binding pocket (encircled by ellipse) that is distinct from the V617 amino acid commonly mutated in myeloproliferative neoplasms. (From Bercovich D, et al. Mutations of JAK2 in acute lymphoblastic leukemias associated with Down’s syndrome. Lancet. 2008;372:1484-1492, with permission.) (D) CRLF2 overexpression: Flow cytometric analysis of cell surface expression of CRLF2 demonstrates CRLF2 overexpression in ALL cases with the P2RY8-CRLF2 fusion and not in fusion-negative cases; CD19 co-staining was performed to demonstrate selectivity for the leukemic cell population. (Modified from Mullighan C, et al. Rearrangement of CRLF2 in B-progenitor and Down syndrome associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243-1246, with permission.)
Submicroscopic Abnormalities
PAX5 and EBF1
In a landmark study, Mullighan and colleagues performed genome-wide analysis of 242 pediatric ALL patient samples using high-resolution single-nucleotide polymorphism arrays and genomic DNA sequencing. They discovered that precursor B-cell ALL samples on average carried 6.63 copy number alterations, indicating relative genomic stability, but confirming the presence of additional subchromosomal cooperating lesions in ALL cases with known leukemogenic karyotypic abnormalities. Notably, about 40% of cases carried aberrations in genes that regulate B lymphocyte development, including EBF1 and PAX5. 24 EBF1 (early B-cell factor) is known to be a master regulator for B-cell development, as Ebf1-deficient mice produce only B-biased progenitor cells but not mature B cells. In addition, EBF1, by remodeling chromatin at the PAX5 locus, is required for PAX5 expression; in turn, PAX5 controls the commitment of the common lymphoid progenitor to the B-cell pathway by repressing genes inappropriate for the B lineage and by activating genes required for B-cell maturation 25 . All known PAX5 aberrations were shown to reduce the transcriptional activity of PAX5 and its downstream targets. However, neither EBF1 nor PAX5 lesions demonstrate prognostic significance. 26
IKZF1, CRLF2, JAK, IL7R, and the “BCR-ABL1-like” Signature
Recurrent deletions and inactivating mutations in the B-cell development gene IKZF1 were also identified by Mullighan and associates. 24,26 IKZF1 encodes the zinc finger lymphoid transcription factor IKAROS (see Figure 26-3, B). Isoforms of IKZF1, which lack N-terminal zinc fingers but retain the ability to dimerize, act as dominant negative inhibitors of IKAROS function and have been shown in murine models to be leukemogenic. Although thought to exist in only 7% of pediatric and 19% of adult ALL patients, the incidence is higher in high-risk ALL populations (29%)—especially in those carrying the t(9;22) Philadelphia chromosome, where they are present in over 80% of pediatric and 63% of adult patients. 27 In addition, IKZF1 deletions are observed at the progression of CML to lymphoid blast crisis (but not in CML chronic phase) and are therefore thought to be central in the pathogenesis of BCR-ABL1 lymphoid leukemia. It was also noted that IKZF1 mutations were highly correlated with (but not pathognomonic for) a “BCR-ABL1-like” gene expression signature even in IKZF1 mutant ALL lacking the t(9;22). 26,28,29
Several other recently identified mutations also give rise to a “BCR-ABL1-like” signature: CRLF2 alterations, JAK mutations (see Figure 26-3, C), and IL7R mutations. 29 CRLF2 forms part of a heterodimeric complex with the interleukin-7 receptor alpha (IL7RA) to serve as the type I cytokine receptor for thymic stromal lymphopoietin (TSLP), a ligand that mediates B-cell precursor proliferation and survival through its activation of downstream JAK/STAT and PI3K/mTOR pathways. Several alterations of CRLF2 have been noted in ALL, all of which lead to CRLF2 overexpression, including a focal interstitial deletion of the pseudoautosomal region of the sex chromosomes that creates the P2RY8-CRLF2 fusion, a translocation of CRLF2 to the immunoglobulin heavy-chain transcriptional enhancer (IGH@-CRLF2) and the CRLF2 F232C point mutation (see Figure 26-3, D). 30–32 CRLF2 alterations have been noted in 5% to 8% of pediatric 29 and 10% to 15% of adult ALL, and with a higher incidence in high-risk ALL, in patients of Hispanic/Latino ethnicity, and in patients with Down syndrome (DS). IL7R gain-of-function mutations are seen in 6% to 10% of ALL and are noted in both B and T phenotypes. 33 CRLF2 aberrations generally occur in cases lacking classic cytogenetic abnormalities, demonstrate a “BCR-ABL1-like” gene expression signature, and tend to co-occur with IKZF1 and/or JAK mutations. 27,34 CRLF2 has been shown to be a negative prognostic indicator in patients with NCI high-risk disease but not in those with standard-risk disease or in those with DS.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


