SEQUENCING
Chung-Han Lee
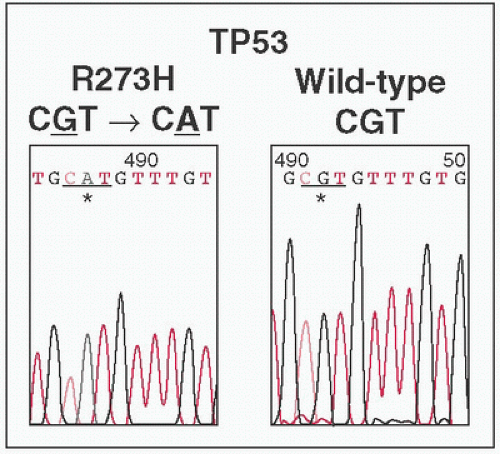
Peaks represent abundance of fluorescent readout at specific nucleotide.
Each dNTP labeled a different color (not shown)
Discrimination of nucleotide identity requires peak to be significantly higher than baseline.
Pure samples yield single peaks at specific location.
Heterogenous mixtures will yield multiple color peaks at specific site (eg, 50% of tumor sample carries point
Mt → tumor:
WT at 1:1 ratio → two different color peaks at 1:1 ratio)
Detection of low frequency point Mts technically difficult (eg, 10% of tumor samples carry point
Mt → tumor:
WT at 1:9 ratio → two different color peaks at 1:9 ratio, smaller peak difficult to distinguish from baseline noise)
Next Generation Sequencing: Other Concepts
Seq by synthesis
DNA is extracted → fragmented → attached to a surface → amplified
Labeled nucleotides are added & detected w/a surface scanner
Labeled nucleotides either replaced by nonlabeled nucleotide or label is removed
Process is repeated w/new labeled nucleotides
Seq will become out of phase w/each successive round → ↓ accuracy + signal
Seq by ligation
DNA is extracted → fragmented → adapters are added & attached to beads & surface → DNA amplified
8-mers w/degenerate primers attached to template
Base 1-2: Specific nucleotides that match template
Base 3-5: Degenerate nucleotides that match multiple things
Base 6-8: Degenerate nucleotides + fluorophore
Detection of fluorophore
Cleavage & removal of bases 6-8 of 8-mer
Repeat w/new 8-mers × 5-7 cycles
Remove all annealed 8-mers & repeat process but offsetting start site by 1 position
End result—seq 35 bps twice (extending w/8-mers 7 times × 5 different start sites × 2 different nucleotides assayed each time)
Each nucleotide assayed on template both as position 1 & position 2 of 8-mer ie, how many times it was seq (Nat Biotechnol 2009;27:1013)
Approaches and Clinical Application of Next Generation Sequencing
Issues illustrated
3
NGS seq projects → 3 sets of Mts discovered
Whole genome seq ≠ whole exome seq
Whole exome seq ≠ targeted exome seq
Cost → use of next generation sequencing (
NGS) for discovery → targeted seq (single
gene) for validation
Next Generation Sequencing: Challenges
Clinical:
How do we determine what
info is clinically relevant? How do we interpret the data that is obtained? Can we predict natural
hx, response to
Rx, or resistance? How do we deal w/tumor heterogeneity? Are
met sites genetically different from
1° sites? Does selective pressure by prior
Rx change a tumor’s genetic profile? Is early detection of resistance genes clinically relevant?
Logistic:
W/c seq technology do we use? What depth of coverage is necessary? What turnaround time for the assay is necessary to be clinically useful? How do we store the massive amounts of data generated? Who is in charge of safeguarding the
info? How do we reduce the costs of the assay? How do we detect Mts that develop w/selective pressure?
Ethical:
GENE EXPRESSION PROFILING
Chung-Han Lee
Gene Expression Profiling
DNA Microarrays
AKA: Gene chips
The principle:
Probes = short unique seq of DNA designed to bind specific cDNA or mRNA
Targets = cDNA or mRNA from genes of interest
A microfluidic chip is designed w/DNA probes placed at known locations
Samples are placed on chip to allow probes to capture targets
Binding of probe & targets are assayed
Data Analysis
Controversial & still subject to research
Fold change as cutoff: Easiest, but arbitrary lacks biologic rationale
Statistical testing such as ANOVA: Complicated by large numbers of genes involved, (eg, p-value < 0.01, examining 10000 genes → 100 by chance alone)
Q-value: Proposed by Yoav Benjamini & Yosi Hochberg, analogue of p-value in
FDR statistical test, helps balance tradeoff between power & error