RESISTANCE TO β-LACTAM AGENTS
β-lactam agents include penicillins, cephalosporins, monobactams, and carbapenems. These agents bind to penicillin-binding proteins (PBPs), which are enzymes (e.g., transpeptidases) involved in the synthesis and maintenance of the peptidoglycan cell wall of gram-positive and gram-negative bacteria. β-lactam resistance can be conferred by decreased access to the PBPs through efflux or outer membrane porin mutations, reduced PBP affinity for β-lactams, and by the expression of β-lactamases.
As previously referenced, staphylococcal β-lactamases are narrow-spectrum penicillinases with relatively poor activity against semisynthetic anti-staphylococcal penicillins like methicillin and oxacillin, cephalosporins, and carbapenems. On the other hand, the majority of β-lactam resistance in clinically relevant gram-positive bacteria (e.g., streptococci, enterococci, and staphylococci) is secondary to expression of low-affinity PBPs (altered drug target).
Methicillin resistance in
S. aureus (MRSA) is due to the expression of PBP2a (
17). This low-affinity PBP is encoded by the
mecA gene. This gene has been mapped to mobile chromosomal elements referred to as staphylococcal chromosomal cassettes (SCC
mec) (
12,
18). Eleven variants of this element have been described (
19,
20,
21,
22). The sizes of these chromosomal cassettes vary, and smaller cassettes (e.g., SCC
mec IV and SCC
mec V) appear to carry fewer concomitant resistance genes (
23,
24). These cassettes contain the
mec gene complex as well as unique cassette recombinase genes (
ccr gene complex) responsible for excision and integration (
18). Contemporary SCC
mec typing is based on variations in these components (
19).
Similarly, penicillin resistance among
Streptococcus pneumoniae is due to the expression of low-affinity PBPs from mosaic gene products (modifications in genes for PBP2b, 2x, and 1a). These mosaic genes are derived from DNA recombination between chromosomal PBP genes and those from less virulent streptococcal species (
25,
26). Ampicillin resistance in enterococci can be due to
β-lactamase production (
27), but high-level ampicillin resistance among
Enterococcus faecium is the result of the expression of a low-affinity target, PBP5 (
28,
29,
30). Mosaic genes resulting in altered PBPs are also responsible for penicillin and cephalosporin resistance among
Neisseria gonorrhoeae (
31,
32,
33,
34).
In gram-negative bacilli,
β-lactam resistance primarily results from the expression of
β-lactamases (
Table 16.1). This heterogenous group of enzymes efficiently hydrolyze the amide bond of the
β-lactam ring, thus inactivating the
β-lactams. These enzymes can be intrinsic (e.g., AmpC
β-lactamases in
Enterobacter cloacae and
P. aeruginosa) or acquired (e.g., plasmid-mediated), and over 1,000 unique
β-lactamases have been described with different structures and spectra of activity (http://www.lahey.org/studies/). These enzymes are classified by two different schemes: the Ambler molecular classification, based on the amino acid structure (
35), and the the Bush-Jacoby-Medeiros functional classification scheme, based on the substrate and/or inhibitor profile (
36). For the sake of simplicity, we will reference the Ambler classification scheme here.
Resistance to ampicillin among
E. coli and
Klebsiella pneumoniae is mediated by narrow-spectrum penicillinases (e.g., TEM-1 in
E. coli and SHV-1 in
K. pneumoniae) (
37,
38). The clinical introduction of third-generation cephalosporins in the 1980s, which were stable against these penicillinases, was soon followed by the description of
β-lactamases capable of inactivating these broad-spectrum antibiotics. Remarkably, a single point mutation in the genes encoding the narrow-spectrum penicillinases was responsible for expanding the substrate spectra (
39). These group of enzymes were designated as extended-spectrum
β-lactamases, or ESBLs (
40). The expression of ESBLs confers resistance to penicillins, first-, second-, and third-generation cephalosporins, and aztreonam. In the absence of other resistance mechanisms, cephamycins and carbapenems retain activity. Interestingly, ESBLs are readily inhibited by
β-lactamase inhibitors (e.g., clavulanic acid). This property has been an important characteristic used for detection of ESBL production in clinical microbiology laboratories. Clinically, however, ESBLproducing
K. pneumoniae and
E. coli tend to be resistant to available
β-lactam/
β-lactamase inhibitor combinations (
41,
42).
Recently, there has been a dramatic shift in the prevalence and types of ESBLs identified both in the community and in many health care settings. The CTX-M family of
β-lactamases, particularly CTX-M-15, have become the dominant ESBL type in Europe, North and South America, and Asia (
43,
44,
45,
46). Unlike SHV-type (e.g., SHV-2) and TEM-type ESBLS, which evolved primarily from mutations in plasmid-mediated penicillinases, CTX-M type ESBLs appear to have evolved from a chromosomal cephalosporinase of
Kluyvera spp. (
47). CTX-M-15 has been associated with a specific clone of
E. coli, ST131 (
44,
48,
49,
50,
51), that has contributed to the successful global dissemination of this particular resistance determinant. CTX-M type enzymes were considered rare in the United States a few years ago, but contemporary surveys demonstrate that its prevalence is increasing at an impressive rate (
52,
53). Many plasmids carrying ESBLs also harbor genes associated with fluoroquinolone and/or aminoglycoside resistance (
44,
54).
Carbapenems are considered the drug of choice to treat patients at risk for or with documented severe infections caused by ESBL-producing organisms, even in the setting of reported
in vitro susceptibility, due to clinical failures observed with alternative therapy (
55).
Phenotypic carbapenem resistance in
Enterobacteriaceae was initially considered sporadic, but during the last decade has become alarmingly more common. Carbapenem resistance results from one or more of the following mechanisms: derepression and hyperproduction of AmpC
β-lactamases or ESBLs with concomitant alteration in outer membrane porins, augmented drug efflux, alterations in PBPs, and/or carbapenemase production (
56).
Currently, most carbapenem resistance among
Enterobacteriaceae in the United States and Israel is attributed to plasmid-mediated expression of a KPC-type (
K. pneumoniae carbapenemase) carbapenemase. These Class A serine carbapenemases hydrolyze carbapenems as well as penicillins, cephalosporins, and aztreonam and are not overcome
in vitro by clinically available
β-lactamase inhibitors (
57). KPCs have been identified in several species of
Enterobacteriaceae as well as
Pseudomonas spp. and
A. baumannii (
56), with KPC-2 and KPC-3 being most commonly isolated subtypes in
Enterobacteriaceae. The
blaKPC gene has been mapped to a Tn
3 based transposon, Tn
4401 (
58), explaining the efficient transfer of these
β-lactamases between strains and species. Five isoforms of Tn
4401 have been described (
59,
60). Reports of many of the plasmids harboring
blaKPC include genes conferring resistance to fluoroquinolones and aminoglycosides are unsettling and not uncommon (
61,
62).
KPC-production confers variable levels of carbapenem resistance with reported minimum inhibitory concentrations (MICs) ranging from susceptible to ≥16 µg/mL. Analysis of 14 KPC harboring
K. pneumoniae isolates with MIC ≥16 µg/mL demonstrated that high level of resistance may be secondary to increased gene copy number (i.e., dose response) or the loss of a functional outer membrane porin, OmpK35 and/or OmpK36. The highest level of resistance was seen with isolates lacking both porins and with augmented KPC enzyme production (
59).
Carbapenem resistance among gram-negative bacilli also can be mediated by the Class B
β-lactamases, the metallo-
β-lactamases (MBLs) (
63). These enzymes use metal, commonly zinc, as a cofactor for
β-lactam hydrolysis. With the exception of aztreonam, MBLs can hydrolyze all
β-lactam antibiotics and are not inhibited by commercially available
β-lactamase inhibitors. Co-expression of MBLs with ESBLs, AmpC
β-lactamases, and/or other carbapenemases can result in concomitant resistance to aztreonam.
Intrinsic carbapenem resistance in
Stenotrophomonas maltophilia is due to chromosomal MBLs. Initially these MBLs were described in
Pseudomonas spp. but reports in
Enterobacteriaceae are now quite common. Frequently detected MBLs include the IMP-type (active against imipenem) and VIM-type (Verona integron-encoded MBL), with the most prevalent being VIM-2. As of 2009, international attention has turned toward the increasing recovery of NDM-1, that is, New Delhi MBL. Due to the conveniences of travel and medical tourism, NDM-1 is not only endemic to the Indian subcontinent, this relatively novel MBL has disseminated worldwide (
64,
65,
66,
67,
68).
The rapid spread of NDM-1 exemplifies the fluidity of gene transfer between bacterial species. Although
blaNDM-1 was initially and repeatedly mapped to plasmids isolated from carbapenem-resistant
E. coli and
K. pneumoniae, reports of both plasmid and chromosomal expression of
blaNDM-1 has been noted in other species of
Enterobacteriaceae as well as
Acinetobacter spp. and
P. aeruginosa (
56,
66). It is currently held that
blaNDM-1 is a chimeric gene that may have evolved from
A. baumannii (
69).
A small percentage of the Class D
β-lactamases, commonly referred to as oxacillinases, demonstrate low-level carbapenemase activity. These carbapenemases contribute to the carbapenem resistance in
Acinetobacter spp. and can be chromosomal or plasmid-mediated. Examples of acquired Class D carbapenemases include OXA-23, OXA 24/40, OXA-58, and
OXA-48. OXA-23 (originally ARI-1) was the first of these types of carbapenem-hydrolyzing oxacillinases described (
70). Both OXA-23 and OXA-58 are responsible for the carbapenem resistance reported in many isolates of
A. baumannii recovered from skin and soft tissue infections in military and civilian personnel returning from the military operations in the Middle East and Afghanistan (
71). OXA-48 appears to have the highest affinity for carbapenems of this class of carbapenemases and has been described with increasing frequency in Turkey, the Middle East, and Europe (
72). High-level carbapenem resistance is usually conferred by the concurrent presence of other resistance determinants including alterations in porins (e.g., CarO in
A. baumannii) (
73), modifications in PBPs, increased transcription mediated by insertion sequences, increased gene copy number, and amplified drug efflux (
74).
Carbapenem resistance in
P. aeruginosa is frequently due to a variety of mechanisms including expression of carbapenem-hydrolyzing
β-lactamases. Other contributors to the carbapenem-resistant phenotype in this species include efflux and alterations in outer membrane proteins. Unlike carbapenemases that, in general, do not discriminate between carbapenems, upregulated expression of certain multidrug efflux pumps (e.g., MexAB-OprM) found in
P. aeruginosa appear to exclude imipenem but confer resistance to meropenem (
3,
75). The loss or alteration of the outer membrane porin OprD, however, is specific for imipenem resistance exclusive of other
β-lactams (
76).
 Occupational Health Services
Occupational Health Services
 Epidemiology and Control of Healthcare-Acquired Infections in Limited-Resource Settings
Epidemiology and Control of Healthcare-Acquired Infections in Limited-Resource Settings
 The Intensive Care Unit, Part A: HAI Epidemiology, Risk Factors, Surveillance, Engineering and Administrative Infection Control Practices, and Impact
The Intensive Care Unit, Part A: HAI Epidemiology, Risk Factors, Surveillance, Engineering and Administrative Infection Control Practices, and Impact
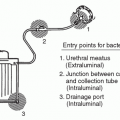 Urinary Tract Infections
Urinary Tract Infections
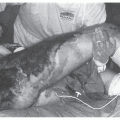 Infections of Burn Wounds
Infections of Burn Wounds



