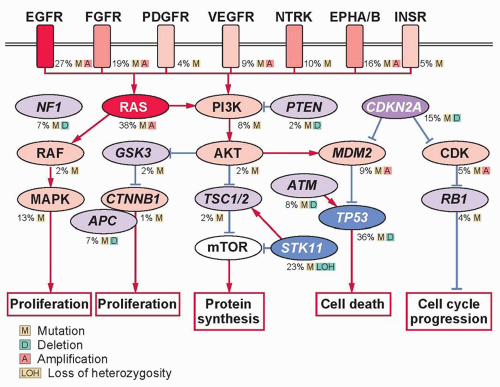
Advances in the understanding of lung carcinogenesis have yielded to a rapid discovery of novel driver oncogenes activated by mutations, translocations, or gene amplification. These alterations occur in genes that encode receptors or intracellular proteins crucial for cellular growth, proliferation, and survival, driving tumor formation and maintenance.
Receptor Tyrosine Kinases
EGFR is one of the most commonly mutated proto-oncogenes in lung adenocarcinoma. It is a transmembrane receptor-tyrosine kinase that is normally activated by binding with one of its ligands, members of the EGF family. EGFR-sensitizing mutations lead to constitutive activation of the tyrosine kinase and phosphorylation of downstream pathways ultimately resulting in uncontrolled proliferation, invasion, and metastasis. In 2004, investigators first noted mutations within the tyrosine-kinase domain of
EGFR in patients who had sustained dramatic responses to the EGFR tyrosine-kinase inhibitors (TKIs), erlotinib and gefitinib.
33,34,35 The frequency of such mutations varies from approximately 10% of lung adenocarcinomas in North American and European populations to as high as 50% in Asia.
36,37,38,39 The leucine to arginine substitution at position 858 (L858R) in exon 21 and short in-frame deletions in exon 19 are the most common sensitizing mutations, comprising approximately 90% of cases. Sensitizing
EGFR mutations are both prognostic for longer survival irrespective of therapy and predictive of response to EGFR TKIs.
35,39,40,41,42,43,44,45,46,47 Other mutations have been described within the
EGFR gene, and for some, the responsiveness to EGFR TKI-directed therapy is similar to the deletion 19 and L858R, including the L861Q and the G719X mutations. An
EGFR sequencing analysis of more than 1,000 patients with adenocarcinoma showed that 27 patients (2.5%) carried exon 20 insertions, representing approximately 9% of all
EGFR mutations.
48 These mutations are found in patients who share the same phenotype as the canonical sensitivity mutations within exon 19 and 21; however, treatment with EGFR TKIs does not result in dramatic responses, and the prognosis is more analogous to that of patients with wild-type
EGFR. A major area of investigation has been the mechanisms of acquired resistance to EGFR TKIs. Both in vitro as well as rebiopsy studies from patients who have been
treated and subsequently progressed on treatment have shown two broad resistance mechanisms, secondary
EGFR alterations and non-
EGFR bypass mechanisms.
49 Approximately 50% of EGFR TKI resistance is due to a second site mutation, the T790M mutation occurring within exon 20.
50,51 Other, non-EGFR mechanisms of TKI resistance include
MET amplification,
PIK3CA mutations, and change in histology to a small cell appearance.
49 The T790M mutation is also found to be present at a very low prevalence in the germ line and appears to increase lung cancer risk independent of smoking.
52ERBB2 (formerly human epidermal growth factor receptor 2 [HER2]/neu) is a member of the erbB receptor family, along with
EGFR. It is overexpressed in about 20% of NSCLCs, and amplified in 2% to 4% of cases.
53 ERBB2 mutations have been identified in approximately 2% of NSCLCs, comprising in-frame insertions of 3 to 12 base pairs into exon 20, usually around the codon 776.
34,38,54 A meta-analysis suggested that ERBB2 overexpression is an indicator of poor survival in lung adenocarcinoma,
55 but amplification or mutations do not seem to be prognostic.
55,56 Early trials examining targeted therapies selected patients on the basis of ERBB2 overexpression and were mostly unsuccessful.
57 However, recent data suggest that trastuzumab, a recombinant humanized monoclonal antibody against ERBB2, may be active in
ERBB2-mutant lung adenocarcinomas.
34 In addition, irreversible TKIs targeting ERBB2 are in early clinical trials.
Hepatocyte growth factor receptor (HGFR) is a receptor tyrosine kinase encoded by
MET.
MET amplification has been reported in about 2% of NSCLCs, and has been implicated in up to 20% of cases with acquired resistance to EGFR TKIs.
58 Several therapeutic strategies, including antibodies that target HGFR and anti-HGFR TKIs are under clinical evaluation in NSCLC patients, mostly aiming to prevent or overcome EGFR TKI resistance.
59,60,61Three other receptors have been nominated as potential druggable targets, especially in lung SQCC.
62,63 Insulin-like growth factor receptor 1 (IGFR1) is a tyrosine kinase receptor that activates the MAPK and PI3K/AKT/mTOR pathways, playing an important role in cancer growth and progression.
64 IGFR1 expression is higher in lung SQCC, and gene copy number has been associated with survival.
65 Despite an initial promise, phase III studies of some anti-IGFR1 agents has halted due to excess toxicities, including treatment-related deaths in the experimental arm. Fibroblast growth factor receptor 1 (FGFR1) is a member of the family of fibroblast growth factor (FGF) receptors that are dysregulated in a variety of cancers.
FGFR1 amplification has been described in approximately 20% of lung SQCCs and has been associated with cigarette smoking and worse survival.
66,67 In vitro studies showed activity with anti-FGFR inhibitors in cell lines harboring this feature, and pan-FGFR and multikinase inhibitors are in early phases of clinical investigation. Discoidin death receptor 2 (
DDR2) is a member of the DDR family of transmembrane receptors that signals through SRC and STAT pathways. Mutations in the kinase domain have been reported in nearly 4% of lung SQCCs and were shown to yield malignant transformation in vitro.
68 Interestingly, some FDA-approved agents for chronic myelogenous leukemia seem to be active against the DDR family, including dasatinib, nilotinib, and imatinib.
69
Translocated Genes in Lung Cancer
The anaplastic lymphoma kinase (ALK) fusion protein is an activating oncogenic driver of lung adenocarcinomas occurring in 3% to 7% of all NSCLCs.
70,71 Multiple ALK variants have been identified in lung cancer, and different ALK partners like kinesin family member 5B (KIF5B) have been described.
70,72,73 Similar to
EGFR mutations, ALK fusions occur almost exclusively in adenocarcinomas, specifically with acinar histology, in never or former light smokers.
70,74,75,76 ALK fusions are not found in tumors with mutations in
EGFR or
KRAS, and patients appear to be of younger age compared to those with
EGFR mutant tumors.
76 The gold standard test for
ALK translocations is fluorescence in situ hybridization (FISH), but a variety of methods have been proposed, including immunohistochemistry, reverse transcription polymerase chain reaction (RT-PCR), and next-generation sequencing (NGS). The clinical activity of crizotinib—an oral inhibitor against c-MET, ALK, and ROS1—was impressive in early trials involving patients with the EML4-ALK translocation.
71 In a randomized phase III trial, crizotinib significantly improved progression-free survival (PFS), response rate (RR), and quality of life in comparison to second-line chemotherapy among patients with metastatic NSCLC harboring ALK translocations.
77 In a fashion analogous to that seen with EGFR TKI resistance, initial reports of resistance mechanisms include both
ALK dependent (e.g., secondary mutations within
ALK or amplification of the mutant
ALK allele) and bypass tracks (e.g., the development of secondary
KRAS and
EGFR mutations have been described).
78,79,80,81 Importantly, several second-generation inhibitors are under clinical investigation and have shown promising activity in patients with acquired resistance to crizotinib.
82,83More recently, fusions involving the receptor tyrosine kinases ROS1 and RET have been described in NSCLC, each with oncogenic activity and potential utility as clinical biomarkers. ROS1 belongs to the insulin receptor family and is not normally expressed in the human lung tissue.
84 It has been demonstrated that ROS1 fusions induce autophosphorylation and downstream activation of common growth and survival pathways like mitogen-activated protein kinase (MAPK), STAT3, and PI3K/AKT.
ROS1 rearrangements occur in approximately 1% to 2% of NSCLC patients, mostly in adenocarcinoma histology, younger patients, and never smokers.
85 It has been shown that selective ALK inhibitors are also active in tumors harboring
ROS1 translocations, both in vitro and in vivo.
86 RET rearrangements were only described in NSCLC in 2011, involving KIF5B as a partner.
87,88 Subsequently, CCD6, NCOA4, and TRIM33 have been described as alternative RET fusion partners.
89,90 Similar to
ALK translocations, RET partners contain a coiled-coil domain that functions as a dimerization unit, leading to ligand-independent homodimerization of the fused protein and constitutive activation of the RET kinase activity mediated by autophosphorylation. Like ALK and ROS1, RET fusions are associated with a lack of smoking history and adenocarcinoma histology, with a frequency slightly higher than 1% in all comers.
91 Agents including cabozantinib,
90 sorafenib, sunitinib, vandetanib, and ponatinib are being evaluated in this patient population.
Intracellular Signaling
Because downstream effectors are required for intracellular transduction of incoming growth factors or receptor signals, it is not surprising that proteins important in cytoplasmic signal transduction cascades are also implicated in carcinogenesis. The
RAS gene family—in particular,
KRAS—can be activated by point mutations at codons 12 or 13 in exon 2 in approximately 20% to 30% of NSCLCs.
KRAS mutations more commonly occur in patients with significant smoking history and adenocarcinoma histology.
92 Approximately 70% are G→T transversions, with the substitution of glycine by either cysteine or valine. Similar G→T transversions are observed in the mutated p53 gene, representing similar DNA damage as a result of bulky DNA adducts caused by the polycyclic hydrocarbons and nitrosamines in tobacco smoke. In a meta-analysis of 3,779 lung cancer patients, mutant
KRAS was correlated to a worse prognosis in adenocarcinoma histology, but this link was not confirmed in a prospective study.
93 The poor prognosis has been proposed to be related to the fact that KRAS mutations rarely occur with EGFR mutations, and EGFR mutant tumors have a better prognosis than EGFR wild type. Thus, if KRAS mutations are evaluated only in EGFR wild-type tumors, the adverse prognostic effect of KRAS disappears.
94 Although the oncogenicity of aberrant
KRAS is well established, effective agents that inhibit KRAS have not
reached the clinic. Another strategy has been to target downstream effectors of RAS, like the RAF family proteins and MEK. For instance, sorafenib—a broad-spectrum kinase inhibitor including the RAF family—led to a disease control rate of 79% in the BATTLE trial
95 among patients with either
KRAS or
BRAF mutations. However, these findings were not confirmed in a large phase III trial.
96 The addition of selumetinib (AZD6244), a MEK1/2 inhibitor, to docetaxel significantly improved RR and PFS in a randomized, phase II trial.
97 A phase III study evaluating this combination in patients with KRAS-mutant NSCLC is ongoing. Another MEK inhibitor, trametinib (GSK1120212), also showed promising results in combination with chemotherapy in early phase I/Ib studies.
98,99 However, it is not clear whether these agents have a differential effectiveness according to the
KRAS status, because similar responses have been observed in
KRAS wild-type tumors.
The RAF proteins are serine/threonine kinases downstream of RAS, represented by three isoforms in mammals (ARAF, BRAF, and RAF1 [also known as CRAF]).
BRAF is a known oncogene in lung adenocarcinoma, being mutated in approximately 3% of cases.
100 In contrast to melanoma, where the majority of
BRAF mutations occur at valine 600 (V600E) within exon 15, V600E comprises only half of cases in lung adenocarcinoma. Interestingly,
BRAF mutations are more likely to occur in former/current smokers. Dabrafenib (BRF113928)—a specific V600E BRAF inhibitor—demonstrated a promising RR of 40% in a phase II trial involving patients with lung adenocarcinoma harboring this mutation.
101 Dramatic responses have also been reported with vemurafenib, another specific inhibitor that is U.S. Food and Drug Administration (FDA)-approved for the treatment of melanoma.
102 Apart from
BRAF, recurrent mutations at
ARAF and
RAF1 inhibitory sites were recently described in lung adenocarcinoma. Importantly, these mutations seem to be oncogenic in vitro, and may predict a response to targeted therapies, including sorafenib and MEK inhibitors.
103MAP2K1 (also known as MEK1) is a serine/threonine kinase downstream of RAF. In NSCLC, point mutations have been identified in 1% of cases, comprising three major nucleotide substitutions (Q56P, K57N, and D67N).
104 These mutations tend to be nonoverlapping with other drivers and induce constitutive extracellular signal-regulated kinase (ERK) phosphorylation and cell proliferation in vitro. It is still unknown if these mutations will predict clinical benefit from anti-MEK therapies. Another frequently altered intracellular pathway in lung cancer is PI3K-AKT-mammalian target of rapamycin (mTOR). Phosphatidylinositol 3-kinases (PI3Ks) are lipid kinases that participate in regulating cell growth, proliferation, and survival. They comprise heterodimers formed by an 85-kDa regulatory subunit and a 110-kDa catalytic subunit, the latter being encoded by
PIK3CA.
PIK3CA mutations have been reported in several cancer types, including a frequency of 1% to 4% in lung adenocarcinoma.
105,106 However, a higher frequency of 16% was reported in the lung squamous section of The Cancer Genome Atlas (TCGA),
107 suggesting a predominance in this histologic subtype. The most common hotspots are at codons E542 and E545 in exon 9 (helical domain) and at codon H1047 in exon 20 (kinase domain). As opposed to other drivers,
PIK3CA mutations often coexist with other oncogenic mutations in the case of lung adenocarcinoma and are not associated with smoking status.
108 PIK3CA mutations were shown to induce constitutive pathway activation and cell transformation in vitro
109 and have been associated with poor clinical prognosis.
110 The best strategy to target PI3K and the population to be selected is still a matter of debate.
AKT1 is downstream of PI3K and is a member of a family of serine-threonine kinases. Recurrent mutations within the plekstrin homology/lipid binding domain of
AKT1 at E17K have been described in up to 1% of patients with lung adenocarcinoma.
111 This mutation causes constitutive activation of AKT and leads to cellular transformation in vitro and in vivo.
112Phosphatase and tensin homolog (PTEN) is a lipid/protein phosphatase that is integral to cellular processes and acts as a TSG gene by inhibiting the activity of the PI3K pathway. While germline mutations in
PTEN lead to Cowden syndrome, somatic mutations have been found in patients with lung cancer, as well as epigenetic inactivation.
PTEN mutations may be more common in smokers and those with squamous cancer, and are also seen in patients with SCLC.
113