Chromosomal translocations
Genes
Comments
Involving RARAα
t(15;17)(q21;q21)
PML
DNA binding, cell proliferation
RARα
Transcription factor
t(11;17)(q23;q21)
ZBTB16 (PLZF)
Zn-finger transcription factor
RARα
Transcription factor
t(5;17)(q32;q21)
NPM1
RNA transport/processing
RARα
Transcription factor
t(11;17)(q23;q21)
NUMA1
Nuclear mitotic apparatus
RARα
Transcription factor
del(17)(q21)
PRKAR1A
cAMP-dependent protein kinase type I alpha regulatory subunit
RARα
Transcription factor
t(4;17)(q12;q21)
FIP1L1
FIP1-like 1
RARα
Transcription factor
t(X;17)(p11;q12)
BCOR
BCL6 corepressor
RARα
Transcription factor
t(2;17)(q32;q21)
NABP1 (OBFC2A)
Oligonucleotide/oligosaccharide-binding fold containing 2A
RARα
Transcription factor
Involving core-binding factor
t(8;21)(q22;q22.3)
RUNX1T1
Zn-finger transcription factor
RUNX1
Runt-like transcription factor
t(3;21)(q26;q22)
MECOM EAP
Contains Zn-finger motif
RUNX1
Runt-like transcription factor
t(12;21)(p13;q22)
ETV6 (TEL)
ETS-like transcription factor
RUNX1
Runt-like transcription factor
t(1;21)(p36;q22)
Unknown
Rare in AML and MDS
t(5;21)(q13;q22)
RUNX1
Runt-like transcription factor
t(17;21)(q11;q22)
inv(16)(p13;q22)
CBFβ
Stabilizes CBF binding to DNA
t(16;16)(p13;q22)
MYH11
Smooth-muscle myosin gene
del(16q)
Involving KMT2A (MLL)
t(11;v)(q23;v)
KMT2A (MLL)
Drosophila trithorax homology
Variable
Many partner genes, AF-9 is commonest
Involving nucleoporin genes
t(6;9)(p23;q34)
DEK
Putative transcription factor
NUP214 (CAN)
Nucleoporin
Cryptic
SET
Unknown function
NUP214 (CAN)
Nucleoporin
t(7;11)(p15;p15)
HOXA9
Homeobox gene
NUP98
Nuclear-pore complex gene
inv(11)(p15;q22)
DDX10
DEAD-box putative RNA
NUP98
Helicase
Nuclear-pore complex gene
Involving genes of the ets family
(t16;21)(p11;q22)
FUS (TLS)
EWS-like RNA-binding protein
ERG
ETS-like transcription factor
t(12;22)(p13;q11)
ETV6 (TEL)
ETS-like transcription factor
MN1
Cloned from meningioma
Other translocations
t(8;16)(p11;p13)
KAT6B (MOZ)
Monocytic leukemia Zn finger
CREBBP (CBP)
Transcriptional activation
t(9;22)(q34;q11)
ABL1
Tyrosine kinase
BCR
Unknown function
The advent of additional molecular methods and their application to leukemias furthered our understanding of the molecular pathogenesis of these neoplasms. Methods that contributed greatly included Southern, Northern and Western blot analysis, fluorescence in situ hybridization, various polymerase chain reaction (PCR)-based methods, and Sanger (first generation) sequencing. In general, molecular methods were used initially to assess single genes, or small numbers of genes. More recently, high-throughput methods such as array-based comparative genomic hybridization (aCGH), single nucleotide polymorphism (SNP) arrays, and next (or second)-generation sequencing have resulted in an explosion of new data. It is now clear from the results of studies that have used whole genome, whole exome, transcriptome (mRNA), and targeted sequencing studies that acute and chronic leukemias are commonly associated with a number of somatic gene alterations and that involve key cellular pathways. Examples of involved pathways include transcriptional regulation of hematopoietic cell development, cell signaling related to tyrosine kinases or cytokine receptors, and chromatin modulation. These studies also have uncovered abnormalities in genes previously unknown to be involved in oncogenesis.
Although next-generation sequencing has shown many gene mutations in leukemias, other molecular mechanisms also have been implicated in leukemogenesis. Gene amplification can result in gene overexpression. Numerical gains or losses of chromosomes, such as trisomies or monosomies due to nondisjunction, are detected in a large subset of acute and chronic leukemias. Gene deletions, like those arising from partial chromosomal deletions or unbalanced translocations, can result in tumor suppressor gene inactivation or loss. Hypermethylation (i.e., epigenetic changes) is another mechanism of gene inactivation. Often more than one mechanism is involved, leading to the accumulation of genetic lesions culminating in leukemogenesis, or subsequently contributing to disease progression.
Traditionally, genes implicated in cancer pathogenesis have been divided into oncogenes and tumor suppressor genes. Oncogenes are considered to play an important role in pathogenesis having acquired constitutive activation through translocation or sporadic mutation, whereas tumor suppressor genes are involved in pathogenesis through their inactivation of gene/protein function. Point mutation, causing missense or nonsense mutations resulting in abnormal or truncated protein products, is a common mechanism of tumor suppressor gene inactivation. Although some genes involved in leukemogenesis follow this paradigm (for example, typical oncogenes HOX and EVI-1 and typical tumor-suppressor genes PU.1 and RARα), some genes demonstrate overlapping features of both oncogenes and tumor-suppressor genes (such as CBF, CEBPA, GATA-1, and NPM1), suggesting that this traditional dichotomy is an oversimplification [2–7].
A new concept of gatekeeper genes has been recently introduced to our understanding of hematologic malignancies. This concept was first established in solid tumors (e.g., colon cancer) where inactivation of a particular pathway is thought to be the first genetic alteration in pathogenesis [8]. Several studies suggest that the genetic alterations affecting hematopoietic transcription factors may be the initiating event resulting in the establishment of a malignant clone, but then secondary events are required for disease manifestation. For example, a myelodysplastic syndrome (MDS ) known to precede acute myelogenous leukemia (AML) is often associated with RUNX1 mutations, which might be an initiating event in AML [9]. Additionally, cytogenetic and genetic abnormalities identified in therapy-related MDS and AML might be initiating genetic events in leukemogenesis [10]. Concordant AML in identical twins, which share the initiating clone carrying the same translocation affecting the MLL gene, supports the idea of gatekeepers in leukemias, as does the observation of RUNX1 or CEBPA mutations in hereditary AML syndromes [11].
Some authors have proposed the concept that leukemogenesis requires the collaboration of at least two classes of mutations: class I, which activate signal-transduction pathways and confer a proliferation advantage to hematopoietic cells; and class II, which affect transcription factors and serve primarily to impair hematopoietic differentiation [12]. While some hematologic malignancies appear to follow this scheme, for example, acute promyelocytic leukemia (APL) carries PML–RARα impairing differentiation and many cases also have FLT3 mutations leading to increased proliferation, there are too many exceptions to believe that this concept can be universally applied for all hematologic malignancies.
30.2 Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is an umbrella term for a group of neoplasms composed of immature cells demonstrating differentiation into one of the myeloid lineages. The approach to classification of AML has changed dramatically over the years, from a purely morphologic system used in the French–American–British (FAB) classification [13] to a system that incorporates morphologic, immunophenotypic, genetic, and clinical features as proposed by the World Health Organization (WHO) classification [14]. The WHO classification divides AML in four major categories: (1) AML with recurrent genetic abnormalities, (2) AML with myelodysplasia-related changes, (3) Therapy-related myeloid neoplasms, and (4) AML, not otherwise specified (NOS).
From the point of view of molecular mechanisms, cases of AML with recurrent genetic abnormalities represent biologically unique entities. Cases of therapy-related AML and AML with myelodysplasia-related changes have overlapping biologic and cytogenetic features. The fourth category, which basically recapitulates the older approach of FAB classification, represents a heterogeneous group driven by a large variety of molecular alterations.
30.2.1 AML with Recurrent Genetic Abnormalities
30.2.1.1 AML Associated with t(8;21)(q22;q22.3): RUNX1–RUNX1T1 (AML1/ETO or RUNX1/MTG8)
The t(8;21)(q22;q22.3) has been identified in 8–12 % of de novo AML [14]. Less commonly, t(8;21) has been identified in therapy-related myeloid neoplasms [15]. In adults, de novo AMLs with t(8;21) respond well to AraC-containing chemotherapy with a high rate of complete remission and relatively long survival. However, the presence of t(8;21) in pediatric AML cases may be less predictive of good prognosis [16]. Morphologically, most cases are classified as M2 using the FAB classification although only a subset of M2 cases carry t(8;21). The myeloblasts in these leukemias typically have distinctive cytological features, characterized by abundant evidence of maturation, including single slender Auer rods with tapered ends and salmon-colored cytoplasmic granules (Fig. 30.1) [17].
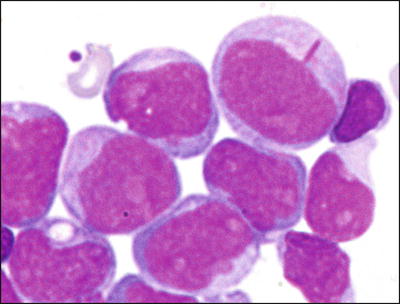
Fig. 30.1
Typical morphology of acute myeloid leukemia with t(8;21). Blasts show single long slender Auer rods with tapered ends. Wright-Giemsa stain, 1000×.
The t(8;21) is a reciprocal chromosomal translocation involving the RUNX1T1 (ETO) gene on chromosome 8 and the RUNX1 (AML1) gene on chromosome 21. As a result of this translocation, the RUNX1T1 and RUNX1 genes are disrupted and fused forming an RUNX1–RUNX1T1 fusion gene, located on the derivative chromosome 8. The RUNX1 and RUNX1T1 genes are fused in a 5′ → 3′ orientation. The fusion protein includes the promoter and runt-like domains of the AML1 protein; the translocation domain of normal RUNX1 is replaced by sequences derived from the RUNX1T1 gene. A reciprocal RUNX1T1– RUNX1 fusion gene located on the derivative chromosome 21 has not been identified. One proposed mechanism for the role of RUNX1–RUNX1T1 in leukemogenesis is competitive inhibition of normal RUNX1 protein. The breakpoints in the RUNX1 gene are consistently detected in intron 5. The breakpoints in the RUNX1T1 gene are also relatively uniform, in the 5′-end of the gene.
The RUNX1 gene encodes for one of many members of a family of heterodimeric transcriptional regulatory proteins. The RUNX1 gene is highly homologous with the Drosophila runt gene, and encodes core binding factor (CBF) α, the human counterpart of the murine nuclear polyoma enhancer binding protein (PEBP2). The binding of RUNX1 protein to DNA occurs via the runt-like central domain and also requires heterodimerization with CBF. The latter protein does not bind to DNA but improves the binding affinity of RUNX1. The RUNX1 gene is normally expressed by myeloid and T-cells and is thought to play an important role in hematopoietic differentiation. The RUNX1T1 gene (also referred to in some studies as CDRor MTG8) encodes a transcription-factor protein that has two zinc-finger-motifs at its C-terminus. The RUNX1T1 gene is expressed in brain, lung, and gonads, but is not normally expressed in hematopoietic cells.
A variety of methods are available to detect t(8;21). This translocation is not difficult to recognize using conventional cytogenetics, which detects t(8;21) in over 95 % of cases (Fig. 30.2). However, rare cases have been reported in which the translocation was not detected by cytogenetics, but was detected by other molecular methods [18]. FISH effectively identifies the t(8;21) (Fig. 30.3). RT-PCR methods detect almost all cases with the t(8;2l). RT-PCR methods, because of their extraordinary sensitivity, also have been used to monitor residual disease. However, studies have shown the persistence of low level RUNX1-RUNX1T1 transcripts in patients in complete clinical remission after chemotherapy or stem cell transplantation, and who have remained in complete remission with prolonged clinical follow-up [19, 20]. Thus, RT-PCR results do not appear to effectively predict risk of relapse after therapy in patients in clinical remission [19, 20].
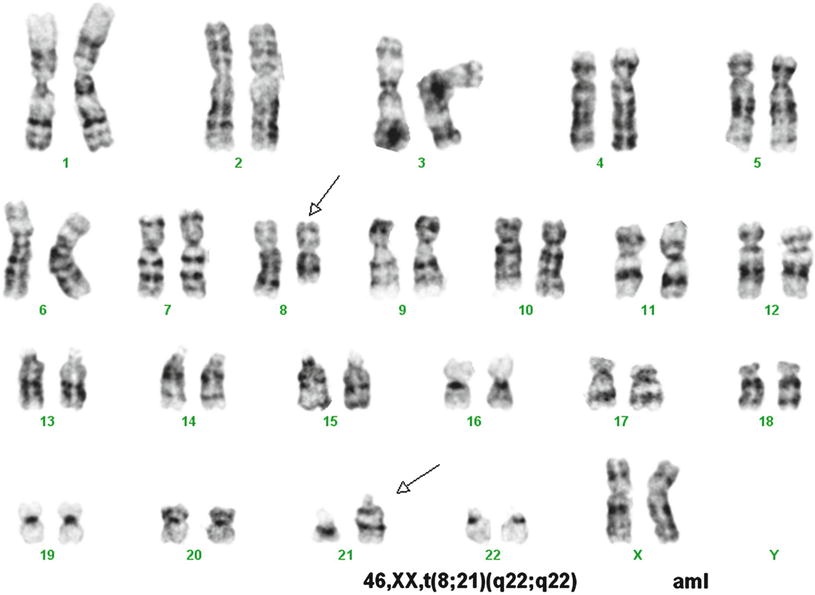
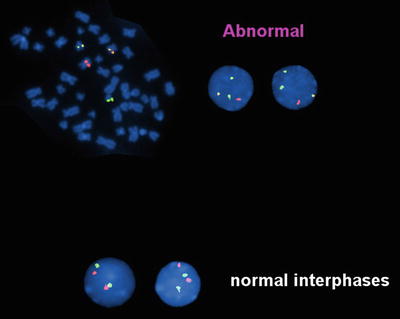
Fig. 30.2
Karyotype of a case of acute myeloid leukemia with t(8;21). Conventional cytogenetic analysis demonstrates t(8;21) as a sole cytogenetic abnormality. GTG banding technique.
Fig. 30.3
Fluorescence in situ hybridization (FISH) analysis of a case of acute myeloid leukemia with t(8;21). Dual color, dual fusion probe (Vysis) in which an RUNX1T1 probe is red and an AML1 (RUNX1) probe is green. In the event of t(8;21) (upper panel) the involved chromosomes show juxtaposition of the red and green probes producing a yellow fusion signal. The uninvolved chromosomes 8 and 21 show red and green signals, respectively. This technique allows analysis of both metaphase (left) and interphase (right) cells. In the absence of t(8;21), cells show two red and two green signal (lower panel).
Cases with RUNX1-RUNX1T1 transcripts appear to follow the class I and class II mutation rule as RUNX1-RUNX1T1 fusions can be detected in long term survivors who are presumably cured of their disease. Knockin mice expressing an RUNX1-RUNX1T1 fusion gene do not develop leukemia until they are exposed to mutagens presumably causing mutations that complement RUNX1-RUNX1T1. Taken together, these findings support the hypothesis that a single genetic event is not sufficient to cause acute leukemia.
30.2.1.2 AML with inv(16)(p13q22) or t(16;16)(p13;q22), (CBFβ/MYH11)
The inv(16)(p13q22) and, more rarely, the t(16;16)(p13;q22) have been identified in 5–12 % of de novo AMLs (Fig. 30.4) [21]. In most cases, the blasts have monocytic cytological features and are associated with immature eosinophils containing coarse basophilic cytoplasmic granules (Fig. 30.5) [21]. The inv(16) also has been identified in rare cases of CML in blast crisis [22].
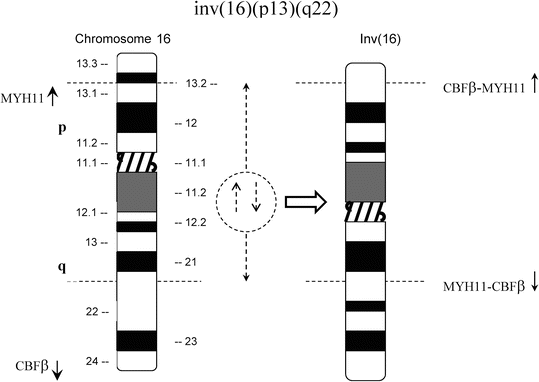
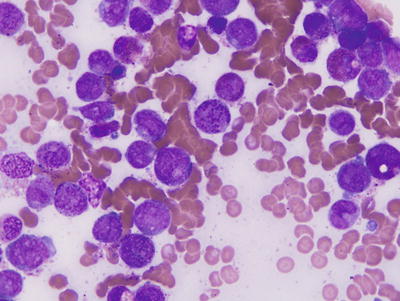
Fig. 30.4
Schematic represent ations of inv(l6)(p13q22) and t(16;16)(p13;q22). Top panel, In inv(l6), most of the CBFβ gene at 16q22 is joined upstream of the 3′ end of MYH11 at 16p13 forming a chimeric CBFβ-MYH11 fusion gene. Bottom panel, The t(16;16) results in a similar chimeric gene on 16p+.
Fig. 30.5
Typical morphology of acute myeloid leukemia with inv(16)(p13q22) or t(16;16)(p13;q22). Immature eosinophilic precursors demonstrate coarse, irregularly sized basophilic granules. Wright-Giemsa stain, 1000×.
The detection of inv(16) in de novo AMLs is a favorable prognostic finding. Patients with AML with inv(16) usually have a higher rate of complete response to chemotherapy, longer duration of remission, and prolonged survival as compared with patients with other forms of AML. The inv(16)(p13q22) involves the CBF (core binding factor or PEBP2) β subunit gene situated on chromosome 16q22 and the MYH11 (smooth muscle myosin heavy chain) gene located at chromosome 16p13. The inversion results in a CBPβ-MYH11 fusion gene from which chimeric mRNA and a novel protein are generated [23]. Both CBFβ and MYH11 are oriented 5′ → 3′ and are transcribed in the centromeric to telomeric direction. The t(16;16) also involves the CBFβ and MYH11 genes, and results in an identical CBFβ -MYH11 fusion gene. A reciprocal MYH11- CBFβ mRNA or protein has not been identified.
The CBF protein has two components, α and β, of which there are three α subunits and one β subunit. The α subunits share a runt domain sequence that allows binding of protein to DNA and the β subunit. The β subunit binds to an α subunit and stabilizes CBF binding to DNA. The normal CBFβ gene spans 50 kb with 6 exons. The breakpoints in the CBFβ gene are relatively constant. In most cases, the breakpoint occurs in intron 5, at nucleotide 495 (corresponding to amino acid 165). However, a small subset of cases with a more proximal breakpoint at nucleotide 399 (amino acid 133) has been reported. The normal MYH11 gene encodes for the smooth-muscle form of the myosin heavy chain, and the gene is a member of the myosin II family [23]. Although the function of the CBFβ-MYH11 protein is not completely known, it is thought to bind to the enhancer or promoters of a number of genes involved in hematopoietic cell differentiation [21]. Gene expression profiling studies of cases of AML with inv(16)(p13q22) have shown upregulation of the NF-ĸB pathway and genes associated with high cell proliferation [24].
The breakpoints in the MYH11 gene are more variable than CBFβ; a number of different breakpoint sites have been reported, although most occur in a small 370 bp intron [21, 24]. This common breakpoint corresponds to nucleotide 1921 in the MYH11 gene. Despite the variability in the different fusion genes generated by the inv(16) and t(16;16), one form of the fusion gene is created in approximately 85 % of cases, involving nucleotide 495 of the CBFβ and 1921 of MYH11 genes. This fusion gene results in the generation of CBFβ-MYH11 protein that includes the first 165 amino acids of the normal CBFβ protein and a relatively small tail portion of the normal MYH11 protein.
Conventional cytogenetic methods, Southern blot analysis, and RT-PCR, have been used to detect the inv(16)(pl3q22) and t(16;16). One major advantage of conventional cytogenetics is that this method will also identify additional chromosomal abnormalities that are present in up to 50 % of cases. However, one potential disadvantage of karyotyping is that the inv(16) can be difficult to recognize or it may be misinterpreted as a del(16). RT-PCR analysis identifies CBFβ-MYH11 transcripts in most cases of AML, with either the inv(16) or the t(16;16), demonstrating that the genetic consequences of the inversion and translocation are identical [22]. Immunohistochemical analysis using an antibody specific for the C terminus of CBFβ-MYH11 also can be used. Cases with inv(16) show nuclear expression, unlike other types of AML [23].
30.2.1.3 Acute Promyelocytic Leukemia (AML with t(15;17)(q24.1;q21.2), PML/RARα and Variants)
The t(15;17)(q24.1;q21.2) occurs exclusively in acute promyelocytic leukemia (APL), classified in the FAB system as M3. APLs represent 5–13 % of all de novo AMLs [26]. There is variation in the genetic predisposition to the development of APL with a higher incidence of APL in adult Latinos compared with non-Latinos in the USA, and a higher incidence in children from Central and South America and Italy [27]. The presence of t(15;17) consistently predicts responsiveness to a specific treatment, all-trans retinoic acid (ATRA). Retinoic acid is a ligand for the retinoic acid receptor alpha (RARα) that is involved in the t(15;17). ATRA is thought to overcome the block in cell maturation, allowing the neoplastic cells to mature and be eliminated. Additional genetic abnormalities are rare in APL suggesting that the t(15;17), by itself, is sufficient for neoplastic transformation. This hypothesis was confirmed by an animal model in which transgenic mice expressing the PML-RARα protein in myeloid cells developed APL and responded to ATRA therapy [28]. Of interest, ubiquitous and unrestricted expression of PML-RARα in embryos is lethal.
Two morphologic variants of APL have been described, typical and microgranular ; both variants carry the t(15;17). In the typical or hypergranular variant (Fig. 30.6) promyelocytes have numerous azurophilic cytoplasmic granules that often obscure the border between the cell nucleus and cytoplasm. Cells with numerous Auer rods in bundles (the so-called faggot cells) are common. In contrast, in the microgranular variant (Fig. 30.6) promyelocytes contain numerous small cytoplasmic granules that are difficult to discern with the light microscope on routinely stained smears, but which are highlighted by myeloperoxidase cytochemical staining and are easily detected by electron microscopy.
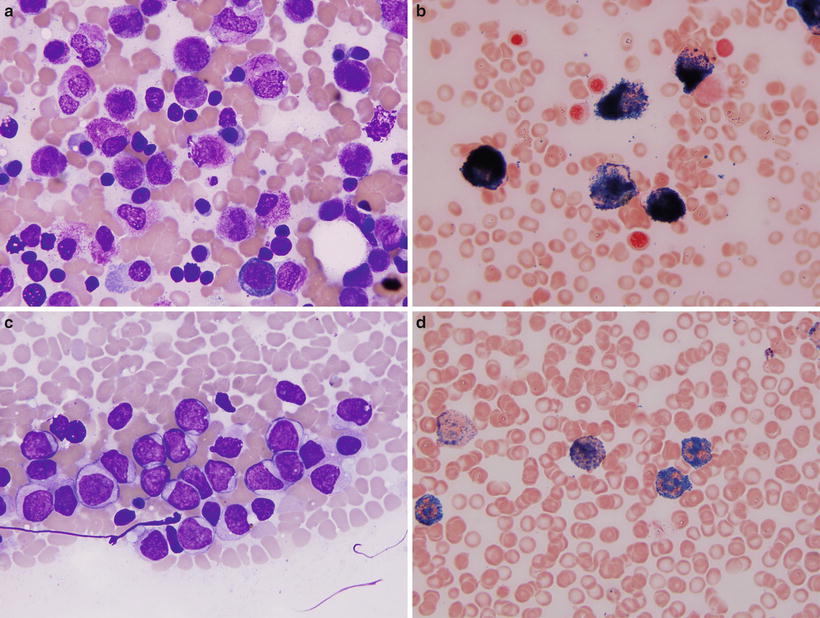
Fig. 30.6
Morphologic variants of acute promyelocytic leukemia. (a, b) Classical variant. Wright-Giemsa stain (a) shows abundant progran ulocytes with numerous granules and occasional bundles of Auer rods (faggot cells). Myeloperoxidase cytochemistry (b) shows numerous positive granules in all neoplastic cells covering entire cell surface. (c–d) Microgranular variant. Wright-Giemsa stain (c) shows immature cells with irregular shaped, convoluted nuclei, and abundant cytoplasm containing small granules that are difficult to appreciate by routine microscopy. However, myeloperoxidase cytochemistry (d) highlights numerous granules similar to the classical variant. Original magnification 1000×.
The t(15;17) is a balanced and reciprocal translocation in which the PML (promyelocytic leukemia) gene on chromosome 15 and the RARα gene on chromosome 17 are disrupted and fused to form a hybrid gene (Fig. 30.7) [29]. The PML–RARα fusion gene , located on derivative chromosome 15, encodes a chimeric mRNA and protein. The PML and RARα genes are oriented in a head-to-tail orientation. The function of the normal PML gene is poorly understood. The gene is ubiquitously expressed and encodes a protein characterized by an N-terminal region with two zinc-finger-like motifs, known as a ring and a B-box, and thought to be involved in DNA binding [29]. A dimerization domain is also present. The normal PML protein appears to have an essential role in cell proliferation. The RARα gene encodes a transcription factor that binds to DNA sequences in cis-acting retinoic acid-responsive elements. High-affinity DNA binding also requires heterodimerization with another family of proteins, the retinoic acid X receptors. The RARα protein, from N-terminal to C-terminal, has transactivation, DNA binding, heterodimerization, and ligand-binding domains. The normal RARα protein plays an important role in myeloid differentiation.
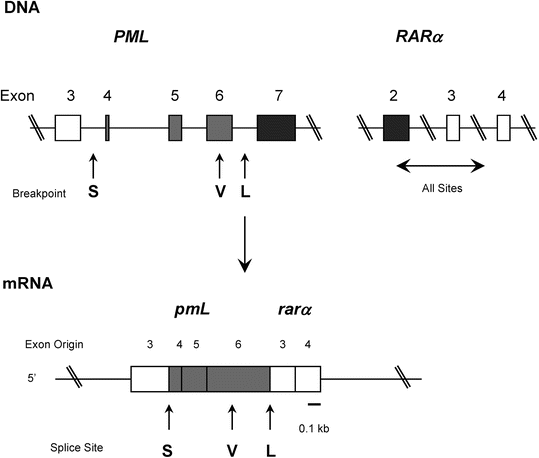
Fig. 30.7
Structure of the PML and RARα genes. S chematic representation of the germ line PML and RARα genes (top), the PML-RARα fusion mRNA transcript (middle), and the long (L) form of the fusion protein (bottom). The lines represent introns, arrows indicate three breakpoints, open boxes represent the exons present in each type of PML-RARα, shaded boxes represent exons excluded from the short (S) form but included in the long (L) or variable (V) forms, and solid boxes indicate exons excluded from all types.
There are three major forms of the PML–RARα fusion gene, corresponding to different breakpoints in the PML gene. The breakpoint in the RARα gene occurs in the same general vicinity in all cases, within intron 2. Approximately 40–50 % of cases have a PML breakpoint in exon 6 (the so-called long form, termed BCR1), 40–50 % of cases have the PML breakpoint in exon 3 (the so-called short form, termed BCR3), and 5–10 % of cases have a breakpoint in PML exon 6 that is variable (the so-called variable form, termed BCR2). In each form of the translocation, the PML-RARα fusion protein retains the 5′-DNA binding and dimerization domains of PML and the 3′-DNA binding, heterodimerization, and ligand (retinoic acid) binding domains of RARα. Recent studies indicate that the different forms of PML-RARα fusion mRNA correlate with clinical presentation or prognosis. In particular, the BCR3 type of PML-RARα correlates with higher leukocyte counts at time of presentation and is more commonly present in the microgranular variant. Higher leukcocyte count and microgranular variant morphology are adverse prognostic findings in APL patients. PML-RARα transcript type does not correlate with survival [30].
In addition to the PML-RARα protein, the t(15;17) results in two other abnormal proteins. An aberrant PML protein is expressed in virtually all cells with the t(l5;17), as a result of alternative mRNA splicing. This PML protein retains its DNA binding capacity and could possibly play a role in neoplastic transformation. A RARα-PML fusion gene is also formed, located on the derivative chromosome 17, and RARα-PML protein is expressed in approximately 75 % of cases of APL. Because this protein lacks the DNA-binding regions of both normal PML and RARα, it is not thought to play a role in leukemogenesis. Furthermore, RARα-PML expression does not correlate with response to ATRA in vitro or with clinical outcome in vivo.
A number of methods can be used to detect the t(15;17). Conventional cytogenetic methods detect the t(l5;l7) in 80–90 % of APL cases at time of initial diagnosis (Fig. 30.8). Suboptimal clinical specimens and poor-quality metaphases explain a large subset of cases with negative results. Fluorescence in situ hybridization (FISH ) (Fig. 30.9) is another useful method for detecting the t(15;17) in APL. Different methods employ probes specific for either chromosome 15 or chromosome I7 (or both), and commercial kits are available. In the past, Southern blot hybridization was used to detect gene rearrangements that result from the t(15;17) but this approach is cumbersome. RT-PCR is another method, convenient and reliable, and detects the various PML–RARα fusion transcripts.
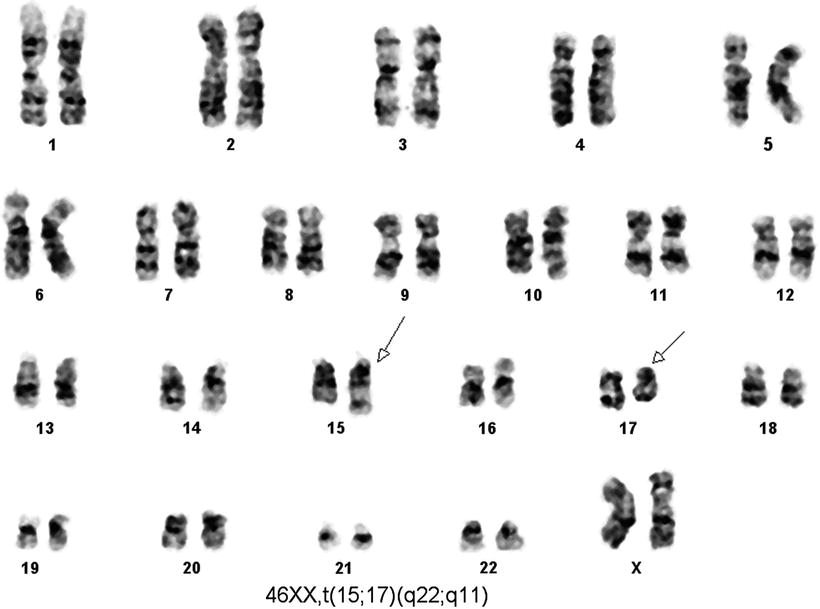
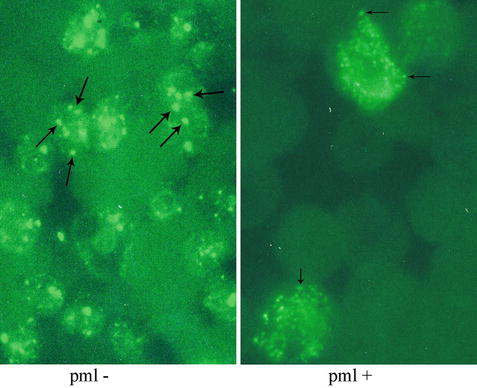
Fig. 30.8
Karyotype of a case of acute promyelocytic leukemia. Conventional cytogenetic analysis using G banding by using trypsin and Giemsa stain (GTG banding technique) demonstrates t(15;17) as a sole cytogenetic abnormality.
Fig. 30.9
Immunofluorescence study using an antibody specific for PML is used to highlight cellular PML oncogenic domains (POD). In the absence of t(15;17), a chunky granular or punctate pattern is observed (left). In the presence of t(15;17), a microgranular pattern is observed (right).
Polyclonal and monoclonal antibodies reactive with the PML and RARα proteins have been generated and immunohistochemical studies assessing the pattern of staining are useful for diagnosis [29, 31, 32]. In normal cells, PML protein is localized in 5–20 spherical structures per nucleus, known as PML oncogenic domains (POD) that are highlighted in a punctate pattern of staining by the anti-PML antibody (Fig. 30.10). RARα protein is not present in these structures. In contrast, in APL cells immunostaining for either PML or RARα reveals a microgranular pattern. Thus, the PML-RARα fusion protein is present in the microgranules. The fusion protein may prevent PML from forming normal PODs because treatment with ATRA allows PML reorganization into these domains.
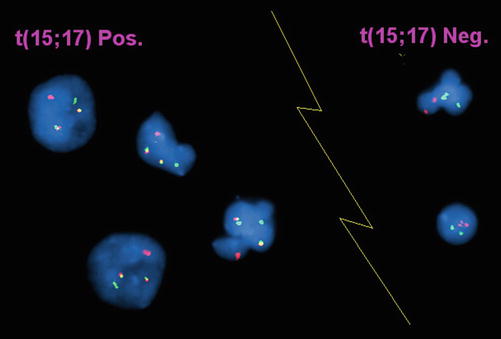
Fig. 30.10
Interphase FISH of acute promyelocytic leukemia.Dual color, dual fusion probe (Vysis) in which the PML probe is red and the RARα probe is green. In the event of t(15;17) (4 cells to the left of the image) the involved chromosomes show juxtaposition of the red and green probes producing a yellow fusion signal. Uninvolved chromosomes show red and green signals, respectively. In the absence of t(15;17), cells show two red and two green signals (2 cells to right of the image).
For the diagnosis of residual disease or early relapse after therapy, conventional cytogenetic studies, Southern blot analysis, and immunohistochemical methods are limited by low sensitivity. In contrast, RT-PCR and FISH methods are very useful. The sensitivity of RT-PCR, which can detect one cell with the PML–RARα gene in 1 × 105 benign cells, makes this method most useful for monitoring residual disease after therapy. For the first few months after therapy, RT-PCR results may be positive with no correlation with relapse rate. However, longer than 3 months after therapy a positive RT-PCR result significantly correlates with an increased rate of relapse [30]. FISH methods offer an advantage over RT-PCR as FISH studies may detect variant forms of the t(15;17) that are negative by RT-PCR. However, several bona fide PML/RARα APL cases with negative FISH results and positive PCR results have been reported in the literature [32]. These cases stress the role of morphological assessment in the establishing the diagnosis of APL and also illustrate that a negative FISH result does not eliminate the diagnosis of APL [32].
t(11;17)(q23;q21)
The t(11;17)(q23;q21) has been rarely identified in AML, in <1 % of all cases. Many patients with t(11;17)-positive AML present with a bleeding diathesis. The blast cells in these cases have cytological features intermediate between the blasts of M2 and the promyelocytes of M3 (FAB). Others have noted that many of these cases have distinctive cup-like nuclei [33]. Unlike APL, patients with t(11;17)-positive AML respond poorly to ATRA.
The t(11;17) is a reciprocal and balanced translocation involving the PLZF (promyelocytic leukemia zinc finger) gene on chromosome 11 and the RARα gene on chromosome 17 [34]. There is some variability in the breakpoints within the PLZF gene. The breakpoints in the RARα gene occur in the second intron, similar to those that occur in the t(15;17). The translocation results in the creation of two fusion genes, PLZF–RARα and RARα–PLZF. Both fusion genes may play a role in leukemogenesis as the structure of both chimeric proteins suggests that either can bind to DNA and influence transcription. The normal PLZF gene is expressed in a tissue specific manner, p articularly in cells of myeloid lineage. PLZF is a member of a large family of zinc-finger transcription factors and is probably involved in myeloid differentiation. The poor ability of ATRA to induce differentiation in t(11;17)-positive AML suggests that disruption of the normal PLZF gene, rather than the RARα gene (as is the case in APL), is the primary leukemogenic event [35]. RARα-PLZF binds to normal PLZF which represses CRABP1 (cellular retinoic acid binding protein 1), thereby resulting in CRABP1 upregulation and thought to be involved in retinoic acid resistance. PLZF-RARα induces MYC expression and may repress normal PLZF in a dominant negative fashion.
Other Rare Variants
Rare partner genes fused with RARα have been identified, greatly aided by the advent of next-generation sequencing methods of cases in which conventional cytogenetics, FISH, and RT-PCR were negative, but the tumors were morphologically typical and had strong myeloperoxidase reactivity. One of the first partner genes was NPM1 (nucleophosmin) in t(5;17)(q32;q21.2). The t(5;17) is a balanced and reciprocal translocation involving NPM1 on chromosome 5 and the RARα gene on chromosome 17. The NPM and RARα genes are fused in a 5′ → 3′ direction. The breakpoint region in the RARα gene is identical to the breakpoint in t(15;17). Other rare gene partners of RARα include: NUMA1 (nuclear mitotic apparatus) [36], STAT5B (signal transducer and activator of transcription 5b) [37], PRKAR1A (cAMP-dependent protein kinase type I alpha regulatory subunit) [38], FIP1L1 (FIP1-like 1) [39], BCOR (BCL6 corepressor) [40], OBFC2A (oligonucleotide/oligosaccharide-binding fold containing 2A) [41], and IRF2BP2 (interferon regulatory factor 2 binding protein 2) (C. Cameron Yin, personal communication).
30.2.1.4 AML with t(9;11)(p22;q23), MLLT3–MLL
The t(9;11)(p22;q23) represents the most common translocation involving chromosome 11q23 in patients with AML and accounts for approximately 2 % of all adult and 9–12 % pediatric AML cases [42, 43]. AMLs with the t(9;11) may be either de novo or therapy-related. De novo neoplasms occur in both children and adults. Patients with de novo AML with the t(9;11) tend to have a more favorable outcome than adults with AML associated with other 11q23 abnormalities. In addition, therapy-related cases of AML have been reported in patients treated previously with chemotherapeutic agents that target topoisomerase II. The t(9;11) is a balanced and reciprocal translocation that disrupts the MLL gene on chromosome 11q23 and the MLLT3 gene (also known as AF9 gene orLTG9 for the leukemia translocation gene) on chromosome 9p22. The translocation results in the formation of an MLL– MLLT3 fusion gene located on the derivative chromosome 11.
The MLL gene (myeloid–lymphoid leukemia or mixed lineage leukemia) gene, also known as ALL1, HRX, or HRTX, is a relatively large gene that is homologous to the Drosophila trithorax gene. MLL is promiscuous and translocations can involve many partners, including MLLT3. MLL consists of 36 exons distributed over 100 kb, and produces a 12 mRNA that encodes a 3968 amino acid protein with an estimated molecular weight of 430 kDa [44]. Recent experiments have indicated that MLL is normally processed via a cytoplasmic cleavage event into a 320 kDa amino terminus (MLL-N), and a 180 kDa carboxy terminus fragment [44]. A number of protein motifs/domains have been identified in the primary structure of MLL, including AT hooks, a DNA methyltransferase domain, PHD domains, a transactivation domain, and a SET domain. An important function of MLL is maintaining HOX gene expression during embryonic development. It has been shown that loss of MLL function in flies and mice results in embryonic lethality and homeotic transformation [44]. The ability of MLL fusion proteins to regulate the expression of HOX genes might be partially responsible for immortalization of myeloid progenitor cells. MLL also influences HOX gene expression via direct binding to promoter sequences [45] and HOXA9 is highly expressed in MLL leukemias [46]. MLL-ENL fusion protein was shown to immortalize immature myelomonocytic cells in vitro. The transplantation of these cells into mice resulted in myeloid leukemia [47]. By fusing a truncated MLL protein to an inducible dimerization domain, Martin et al. have shown that MLL fusion proteins must dimerize to immortalize hematopoietic cells and inhibit their differentiation [48]. Biochemical analysis of MLL suggests that it normally functions as a transcriptional regulator, and expression of MLL fusion proteins has been shown to be leukemogenic in mice.
Despite the large size of the MLL gene, most (but not all) MLL translocations associated with hematologic malignancy can be mapped to an 8.3 kb breakpoint cluster region (bcr). This region encompasses MLL exons originally designated exons 5–11. Subsequent studies revealed that MLL exon 4 actually consists of three discrete exons (now designated exons 4A, 4B, and 4C). There is a correlation between the locations of the breakpoints in de novo versus therapy-related AML. Using the XbaI restriction enzyme, the 8.3 kb fragment of DNA can be divided into a 5′ 4.6 kb region I and a 3′ 3.9 kb region II [49]. De novo AMLs more commonly have breakpoints in region I. In contrast, most cases of therapy-related AML have region II breakpoints [49]. The presence of scaffold-attachment regions and possible topoisomerase II consensus binding sites in region II may explain the increased likelihood of region II breakpoints in therapy-related AML [49].
Conventional cytogenetics is an excellent method to detect 11q23 abnormalities. This technique allows the detection of translocations involving 11q23 and also identifies all possible partner chromosomes. However, a subset of AMLs with 11q23 abnormalities may have a normal karyotype [50]. FISH techniques are also useful, and may detect a small subset of cases not recognized by conventional cytogenetics. The clustering of breakpoints within the MLL gene is well-suited to their detection by Southern blot analysis [51]. Using BamHI digested DNA, a single MLL cDNA probe spanning an 8.3 kb genomic fragment detects most of the common and uncommon 11q23 translocations as gene rearrangements. The introns between exons 5 through 11 are very large and thus standard PCR methods cannot be used for detecting 11q23 translocations. However, the cDNA corresponding to exons 5 through 11 is <700 bp, allowing RT-PCR analysis. A panel of primers needs to be used to detect the l lq23 translocations because of the number of possible partner chromosomes. Multiplex RT-PCR approaches are most convenient.
One of the more unusual features of leukemias associated with MLL fusion is that there is a very brief latency period. Leukemias with MLL fusions have been detected in newborns [52] and even in aborted fetuses [53]. The concordance rate of leukemia in monozygotic twins who have MLL fusions is almost 100 % [54]. It has been hypothesized that additional mutations occur very rapidly in cells that acquire MLL fusions [55]. One possible mechanism is that MLL fusion leads to a mutator phenotype [55]. MLL fusions have been shown to impair DNA double strand break recognition [56] and cell cycle checkpoints [57], both of which can lead to increased accumulation of mutations. It is also possible that the insult caused by MLL translocation can cause additional mutations, especially if the individual had an inherited deficiency in DNA repair or toxin metabolizing enzymes. This possibility is supported by studies that show that certain isoforms of NQO1 or CYP3A4 confer an increased risk of infant leukemia [58] or therapy-related leukemia [59].
30.2.1.5 AML with t(6;9)(p23;q34); DEK-NUP214
The t(6;9)(p23;q34) (Fig. 30.11) occurs in 1–2 % of cases of AML [60]. Patients with AML and the t(6;9) are commonly of young age (<40 years old), and have a poor prognosis [60]. Appro ximately 50 % of patients achieve clinical remission with chemotherapy, but relapse is common. A subset of patients presents with aggressive MDS rather than AML; others present with overt AML with multilineage dysplasia [60].
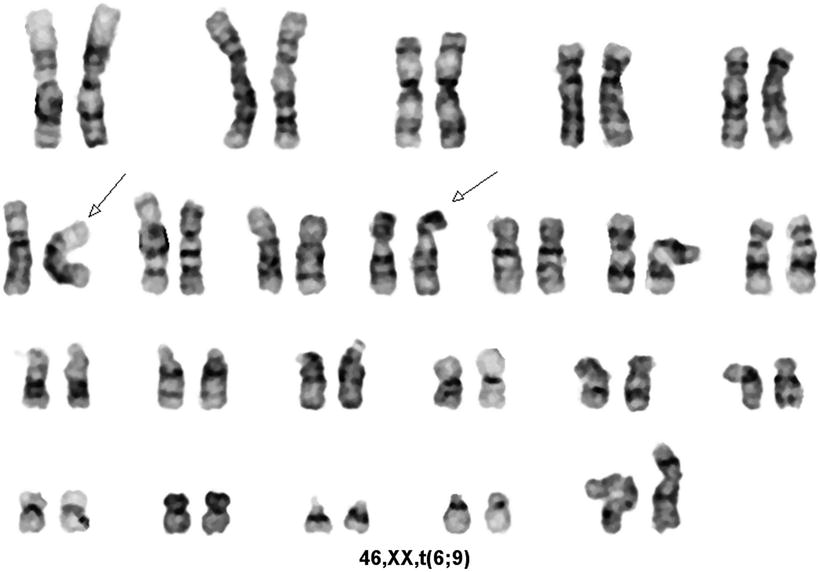
Fig. 30.11
Karyotype of a case of acute myeloid leukemia with t(6;9)(p23;q34). GTG banding technique.
The t(6;9) disrupts the DEK gene at chromosome 6p23 and the CAN (NUP214) gene at chromosome 9q34, resulting in a DEK-CAN fusion gene on the derivative chromosome 6 [61]. The breakpoints in t(6;9) are clustered. The breakpoints in the DEK gene occur in one intron, known as icb-6 (for intron containing breakpoint on chromosome 6). The breakpoints in the CAN gene also cluster in one intron, known as icb-9. The DEK gene is approximately 40 kb in size and encodes a 43 kDa protein, located in the cell cytoplasm, and thought to be a transcription factor. The CAN gene is relatively larger (over 140 kb) and encodes a 214 kDa protein. The normal CAN protein is a component of the nuclear-pore complex, involved in transport of mRNA and proteins between the cytoplasm and the nucleus [61]. The DEK-CAN fusion protein, of predicted size of 165 kDa, has a nuclear distribution, suggesting it is a part of the transcription factor system [61]. It has been suggested that DEK-CAN initiates AML within a subpopulation of hematopoietic stem cells [62].
The t(6;9) is effectively detected by conventional cytogenetics, which also allows the detection of additional abnormalities that may occur with disease progression; trisomies of 8 and 13 are the most common additional abnormalities [60]. The clustering of breakpoints in icb-6 of DEK and icb-9 of CAN also allows convenient detection by using RT-PCR methods. FLT3 gene mutations are common in t(6;9)-positive AML [60, 63].
30.2.1.6 AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1
AML associated with inv(3) or t(3;3) is uncommon, representing 1–3 % of all cases of AML. This disease occurs in adults and men and women are equally affected [64]. De novo AML and cases arising from MDS have been described [65]. Inv(3) or t(3;3) is usually associated with other cytogenetic abnormalities and is rarely a sole abnormality.
Patients with AML with inv(3) or t(3;3) can present with prominent hepatosplenomegaly [65]. Platelet counts in the peripheral blood at presentation may be increased, normal or decreased, and demonstrate a wide range of values, from 20 to 1731 × 109/L [64, 65]. AML with inv(3) or t(3;3) is associated with increased number of micromegakaryocytes and monolobated and bilobated megakaryocytes in the bone marrow (BM). Many cases also show dysplastic changes in the erythroid and/or myeloid lineages [65, 66].
It has been shown that the chromosomal breakpoints in 3q26 are widely scattered in the 5′ or the 3′ regions of the EVI1(Ecotropic viral integration site 1) gene currently known as MECOM(MDS1 and EVI1 complex locus) gene [67], whereas the chromosomal breakpoints in the 3q21 region are restricted to a relatively narrow area. Two different clusters have been identified downstream of the RPN1 (ribophorin I) gene [68]. Alternative splicing of EVI1 results in the creation of MDS1/EVI1 transcripts. Suzukawa et al. suggested that the housekeeping gene RPN1 acts as an enhancer of EVI1 expression, resulting in leukemogenesis [68]. It has been demonstrated that ectopic expression of EVI1 in immature hematopoietic cells interferes with erythroid and granulocytic development [69]. While there is a single report in the literature describing good response of two patients with inv(3) to thalidomide and arsenic trioxide [70], in general, the prognosis of patients with inv(3) or t(3;3) is poor with short survival [66, 71].
30.2.1.7 Acute Megakaryoblastic Leukemia with t(1;22)(p13;q13); RBM15-MKL1
Most cases of AML with t(1;22) have been reported in infants [72]. The largest series published to date describes 39 cases with a m edian age of 4 months (range, 1 day to 36 months) [72]. The t(1;22) is a sole cytogenetic abnormality in more than half of the cases and is a sole cytogenetic abnormality much more often in infants younger than 6 months of age (19 of 23, 83 % of cases), than in older infants (3 of 16, 19 % of cases) [72]. There is a slight female predominance; all patients reported had not had evidence of Down syndrome [72]. In rare cases, RBM15–MKL1 transcripts result from 3-way translocations involving 1p13 and 22q13, such as t(1;14;22) or t(1;22;4) [73]. Almost all patients present with organomegaly [72]; a subset of patients was initially diagnosed with a solid tumor [72]. Moderate leukocytosis with circulating blasts, marked anemia, and variable thrombocytopenia are usually observed at presentation [72, 73]. Most (~90 %) patients in the largest series reported [72] fulfilled the diagnostic criteria for AML M7. Patients usually have a hypercellular or normocellular BM; however, some patients have hypocellular BM [73]. Blasts may exhibit typical morphologic features of megakaryocytic differentiation (i.e., cytoplasmic basophilia, blast clumping, cytoplasmic blebs) or have an undifferentiated appearance [72]. The blasts usually express immunologic markers of megakaryocytic differentiation (i.e., CD42b, CD41a, Factor VIII) [72]. A subset of blasts also expresses CD34 and HLA-DR; some blasts may express the myelomonocytic marker, CD33 [72].
The t(1;22)(p13;q13) results in a fusion of RNA-binding motif protein-15(RBM15) and megakaryocyte leukemia -1 (MKL1) genes [74]. t(1;22)-positive blasts express both reciprocal fusion transcripts, RBM15–MKL1 and MKL1–RBM15, as detected by RT-PCR [74]. However, the predicted RBM15–MKL1 chimeric protein encompasses all putative functional motifs encoded by each gene, which makes it the candidate oncoprotein of t(1;22). Ma et al. suggested in cases with RBM15–MKL1 the MKL1 SAP domain is expected to aberrantly relocalize the RRM and SPOC motifs of RBM15 to sites of transcriptionally active chromatin, deregulating RNA processing and/or Hox and Ras/MAP kinase signaling and altering the normal proliferation or differentiation of megakaryoblasts [74]. The prognosis of patients with AML with t(1;22) is intermediate and appears to have improved dramatically with modern therapy. The largest series published in 1999 reported a 53 % complete remission rate and median survival of 8 months (range, 1 day to 104 months) [72]. A more recent report of 11 pediatric patients published in 2003 described 6 of 11 patients being alive after more than 2 years follow-up [73].
30.2.1.8 AML with Mutated NPM1
The NPM1 (nucleophosmin) gene, mapping to chromosome 5q35, contains 12 exons [74]. It encodes for three alternatively spliced nucleophosmin isoforms: B23.1, B23.2, and B23.3. Nucleophosmin is a highly conserved phosphoprotein that is ubiquitously expressed in tissues [74]. The bulk of NPM resides in the granular region of the nucleolus, but NPM shuttles continuously between nucleus and cytoplasm [74]. NPM, through its involvement in ribosome biogenesis, plays a central role in cell growth and proliferation. NPM1 mutations result in cytoplasmic accumulation of NPM that can be detected by immunohistochemical analysis.
NPM1 functions both as an oncogene and a tumor-suppressor gene depending on gene dosage, expression levels, interacting partners, and compartmentalization [2]. NPM1 appears implicated in promoting cell growth, as its expression increases in response to mitogenic stimuli and above normal amounts are detected in highly proliferating and malignant cells. On the other hand, NPM1 contributes to growth suppressing pathways through its interaction with ARF.
NPM1 mutations are relatively specific for AML [75]. Although rare cases of NPM1 mutations have been described in patients with chronic myelomonocytic leukemia and MDS, many of these patients rapidly progress to overt AML [76]. NPM1 mutations are relatively common in de novo AML, but rare in AML secondary to myeloproliferative/myelodysplastic disorders and therapy-related AML. NPM1 mutations are characteristically heterozygous and retain a wild-type allele. In children with AML, the frequency of NPM1 mutation ranges from 2.1 % in Taiwan to 6.5 % in Western countries accounting for 9–26.9 % of all childhood AML with normal karyotype. In adult AML patients, the frequency of NPM1 mutations ranges between 25 and 35 % accounting for 46–64 % of adult AML with normal karyotype [74, 77–79]. Except for two cases involving the splicing donor site of NPM1 in exon 9 or exon 11 [80], NPM1 mutations are restricted to exon 12. Loss of NPM1 mutations at relapse is rare and may be due either to emergence of a different leukemic clone or inability to detect mutations because few leukemic blasts are infiltrating the BM. AML patients with a wild type NPM1 gene at diagnosis rarely acquire NPM1 mutations during the course of disease, suggesting that mutations are unlikely to play a role in disease progression.
NPM1 mutations are associated with a normal karyotype and do not occur in core binding factor leukemias and acute promyelocytic leukemia [75]. Chromosomal abnormalities observed in 14 % of NPM1 mutated AML cases usually arise in subclones and are probably secondary events associated with clonal evolution. Several studies have shown a strong correlation between NPM1 mutations and FLT3-ITD [77, 78].
NPM1-mutated AML cases show a wide morphologic spectrum, but NPM1 mutations are more frequent in the FAB M4 and M5 categories [78] and in AML with prominent nuclear invaginations (cuplike nuclei) [33]. More than 95 % of NPM1-mutated AML cases are CD34+ [81]. NPM-mutated AML cases with a normal karyotype often have high blast counts, high FLT3-ITD and LDH serum levels, extramedullary involvement (mainly gingiva and lymphadenopathy) and higher platelet counts. Bone marrow in NPM1-mutated AML cases frequently shows an increased number of megakaryocytes exhibiting dysplastic features [82]. After induction therapy, AML with a normal karyotype carrying mutated NPM shows a higher complete remission rate than AML with normal karyotype without NPM1 mutations [83]. NPM1 mutation status, however, is not an independent predictor for responsiveness to chemotherapy [81].
Conventional screening of NPM1 mutations is usually performed by PCR using a fluorescence conjugated primer prior to fragment analysis by capillary electrophoresis. Immunohistochemical analysis to detect abnormal cytoplasmic localization of mutated NPM1 protein is another common screening method [81, 84]. Alternative methods for detecting NPM1 mutation include denaturing high performance liquid chromatography and fluorescence resonance energy transfer technique, and direct sequencing.
30.2.1.9 AML with Mutated CEBPA
The CCAAT/enhancer-binding protein a ( CEBPA ) gene encodes a protein member of the basic region leucine zipper (bZIP) transcription factor family, playing a crucial role in granulopoiesis [85]. There are two main categories of CEBPA mutations: (1) C-terminal mutations that occur in the bZIP domain, which are usually in-frame and predict mutant proteins lacking DNA binding and/or homodimerization activities, and (2) N-terminal nonsense mutations that prevent expression of the full-length protein and result in truncated isoforms with dominant-negative activity. Some patients present with biallelic mutations at the C-terminus, whereas others are heterozygous for separate mutations or have a C-terminal mutation coexisting with a mutation in the N-terminus [86, 87].
CEBPA mutations usually occur in AML with a normal (diploid) karyotype and represent 15–19 % of cases in patients with normal cytogenetics. CEBPA mutations are associated with a favorable prognosis [79, 86, 88], and significantly better event-free survival [87, 89], disease-free survival [89], and overall survival [87, 89]. At diagnosis, patients with CEBPA mutations often have higher percentages of peripheral blood blasts, lower platelet counts, less lymphadenopathy and extramedullary involvement, less frequent FLT3/ITD and FLT3/TKD mutations, and rare MLL/PTD mutations compared with AML patients without CEBPA mutations [86]. Complete remission rates do not differ between patients with and without CEBPA mutations [79, 86, 88], but complete remission duration [86], disease-free survival [88], event-free survival [79], and overall survival [88, 96] are better for patients with CEBPA mutations. On multivariate analysis, CEBPA mutational status adds prognostic information to that provided by MLL/PTD and FLT3/ITD status, age, and resistant disease after the first course of induction therapy with regard to complete remission duration and by FLT3/ITD status, age, and WBC for overall survival [86]. In another study, CEBPA mutations, BAALC expression, and FLT3/ITD status were prognostic for disease-free survival, and CEBPA mutations, age, BAALC expression, and FLT3/ITD status were prognostic for overall survival [88]. Fröhling et al. [86] analyzed clinical outcomes in patients with N-terminal nonsense CEBPA mutations, patients with other CEBPA mutation types, and patients with wild-type CEBPA and found that the complete remission duration was the longest in patients with N-terminal mutations, followed by the complete remission duration in patients with other mutations and in those without CEBPA mutations. However, in pairwise comparisons, there were no significant differences in the complete remission duration between patients with N-terminal mutations and those with other mutations or between patients with other mutations and patients with wild-type CEBPA [86].
30.2.2 AML Associated with Translocations Not Included in the AML with Recurrent Genetic Abnormalities Category of the Current WHO Classification
30.2.2.1 AML with 11q23 (MLL) Abnormalities Other Than t(9;11)
Translocations involving the 11q23 locus are detected in 3–10 % of all patients with de novo AML [90]. In infants (age <1 year), AML with 11q23 translocations typically presents with hyperleukocytosis and has a poor prognosis [91]. Translocations involving chromosome 11q23 are found in more than 70 % of leukemia patients younger than 1 year of age whether the immunophenotype is designated AML or ALL. In adults, AMLs with 1lq23 translocations most commonly exhibit monocytic maturation and are classified as FAB M4 or M5. These AMLs do not have specific clinical features and may have a poorer prognosis than AMLs without 11q23 translocations. In addition, 80–90 % of therapy-related AMLs that occur in patients previously treated with topoisomerase II inhibitors are associated with 11q23 translocations. Translocations involving the 11q23 locus are not restricted to AML and are associated with different hematologic malignancies including precursor T or B lymphoblastic leukemia, MDS, and Burkitt lymphoma [51, 90]. In addition, MLL has been labeled a promiscuous oncogene since translocations involving over 60 partner genes or regions have been identified [90].
AML/MDS with t(11;16)(q23;p13.3). The t(11;16) is a rare recurrent translocation that has been identified in therapy-related AML and MDS, occurring almost exclusively in patients treated previously with agents that inhibit topoisomerase II [92]. The t(11;16) is a reciprocal translocation that disrupts the MLL gene at 11q23 and the CBP cAMP response element or CREB-binding protein) gene at 16p13.3, resulting in the formation of an MLL–CBP fusion gene located on the derivative chromosome 11 [92]. A CBP–MLL fusion gene is also produced, but not in all cases, and therefore is not thought to be leukemogenic. The normal CBP protein is a transcriptional adaptor/coactivator protein.
Other 11q23 translocations in AML. Other 11q23 translocations less commonly identified in AMLs include the t(6;11)(q27;q23), t(11;19)(q23;p13.3), t(11;19)(q23;p13.1), t(1;11)(p32;q23), t(l;11)(q21;q23), t(11;17)(q23;q21), and t(10;11)(p11;q23). In each of these translocations, the MLL gene is disrupted and its 5′ end is fused with the 3′ end of the partner gene: AF6 (MLLT4) in the t(6;11), ENL (MLLT1) in the t(11;19)(q23;19p13.3), ELL in the t(11;19)(11q23;19p13.1), AFlp in the t(1;11)(p32;q23), AFlq in the t(l;11)(q21;q23), AF17 (MLLT6) in the t(11;17), and AF10 (MLLT10) in the t(10;11) [93]. The formation of these fusion genes, all located on the derivative chromosome 11, results in the generation of a novel chimeric protein.
30.2.2.2 11q23 Rearrangements Identified in AMLs with Normal Cytogenetics and Trisomy 11
MLL gene rearrangements have been shown in ~10 % of AML with a normal karyotype. All were shown to have partial tandem duplications (PTD) of the 5′ part of the MLL gene [50]. These mutations occur in 3–10 % of adult AML cases, but are uncommon in pediatric AMLs. MLL-PTDs are extremely frequent in conjunction with trisomy 11 [50, 94, 95] and are associated with an unfavorable outcome [50, 96]. Another type of intrachromosomal rearrangement of the MLL, intrachromosomal amplification, also occurs. This type most likely leads to increased MLL expression in AML and MDS [97].
30.2.2.3 AML with inv(11)(p15q22)
The inv(11)(p15q22) has been identified in a very small number of de novo and therapy-related cases of AML [98]. This inversion disrupts the DDX10 gene at chromosome 11q22 and the NUP98 gene at 11p15, creating the NUP98-DDX10 fusion gene. A reciprocal DDX10-NUP98 fusion gene is also created and may be expressed, but is not thought to be involved in neoplastic transformation [98]. The normal DDX10 gene is large, spanning 200 kb, and is composed of at least 12 exons. This gene is a DEAD-box putative RNA helicase gene that encodes a protein that may be involved in ribosomal assembly [99]. The DDXl0 gene is ubiquitously expressed in normal tissues. The normal NUP98 gene encodes a nuclear-pore complex protein that is also ubiquitously expressed.
30.2.2.4 AML with t(16;21)(p11;q22)
Most neoplasms with the t(16;21) have been AMLs, but rare cases of CML in blast crisis and MDS with the t(16;21) have been desc ribed [100]. Patients with AML associated with the t(16;21) are generally young and have a poor prognosis. The t(16;21) is a reciprocal and balanced translocation that disrupts the TLS/FUS gene at chromosome 16p11 and the ERG (ETS-related gene) gene on chromosome 21q22 [101]. As a result two fusion genes are created, TLS/FUS-ERG on the derivative chromosome 21 and ERG-TLS/FUS on the derivative chromosome 16. Although both fusion genes may be expressed, only the TLS/FUS-ERG fusion gene is consistently expressed in all cases, suggesting that this chimeric protein is involved in leukemogenesis [101]. Both the TLS/FUS and ERG genes are oriented 5′ → 3′. The normal TLS/FUS gene encodes an RNA-binding protein that is highly homologous to the EWS gene involved in Ewing sarcoma. The TLS/FUS protein plays a role in activating transcription. The normal ERG gene, a member of the ETS proto-oncogene superfamily, also encodes an RNA-binding protein and is a potent transcriptional activator [101]. At least four different transcripts of the TLS/FUS-ERG have been identified. These transcripts result from variability in the breakpoints and alternative RNA splicing. In most cases, the breakpoint in the ERG gene is tightly clustered in one intron. The breakpoints in the TLS/FUS gene are more variable [100].
30.2.2.5 AML with t(12;22)(p13;q11)
The t(12;22)(p13;q11) is found in AML and rare cases of MDS and CML. The t(12;22) is a reciprocal translocation involving the ETS variant 6 (ETV6) gene (known previously as TEL oncogene) at chromosome 12p13 and the MNI (meningioma) gene at chromosome 22q11 [102]. The translocation disrupts the ETV6 and MNI genes, resulting in the formation of MNI-ETV6 and ETV6-MNI fusion genes. The MNI-ETV6 fusion gene is likely to encode the leukemogenic protein, based on the predicted structure of the fusion protein, which is consistent with an altered transcription factor. The MNI-ETV6 fusion gene also has been constantly expressed in the small number of neoplasms analyzed, unlike the ETV6-MNI fusion gene [102]. The normal ETV6 gene is very large, exceeding 150 kb, and is a member of the ETS gene family of transcription factors. ETV6 is ubiquitously expressed in tissues. The normal MNI gene is less well-known. The gene was originally cloned from a t(4;22)(p16;q11) identified in a case of sporadic meningioma [103]. The gene spans 70 kb, has at least two exons separated by a large intron, and encodes a protein predicted to have 1319 amino acids. The breakpoints in the MNI gene appear to be clustered in the 5′-region of the intron [103].
30.2.2.6 AML with t(8;16)(p11;p13)
The t(8;16)(p11;13) is identified in <1 % of all cases of AML. Most AMLs with the t(8;16) exhibit monocytic differentiation and are morphologically classified using the FAB as M4 or M5 [104]. The blasts often exhibit evidence of erythrophagocytosis. Both de novo and therapy-related AMLs with the t(8;16) have been reported. De novo cases commonly occur in children and adolescents (<18 years of age) [104]. The t(8;16) is a reciprocal translocation, involving the MOZ (monocytic leukemia zinc-finger protein) located at chromosome 8p11 and CREB binding protein (CREBBP) gene located at 16p13 [105]. The translocation disrupts these genes, resulting in the formation of a MOZ-CBP fusion gene on the derivative chromosome 8. A CREBBP -MOZ fusion gene is also created, but is thought to be nonfunctional. The MOZ gene encodes a 225 kDa protein that is widely expressed in tissues. The protein has both zinc-finger and acetyltransferase domains and may mediate leukemogenesis by aberrant acetylation of chromatin [105]. The CREBBPgene spans 190 kb and encodes a protein involved in transcriptional activation. Mutations in the CREBBP gene have been identified in a rare genetic disease, the Rubinstein–Taybi syndrome, characterized by mental retardation, dysmorphic cranial features, and digital abnormalities [106].
30.2.2.7 AML with t(3;21)(q26;q22)
The t(3;21)(q26;q2.2) has been reported in cases of chronic myelogenous leukemia (CML) in blast crisis, in MDS, and in AML following treatment with topoisomerase II inhibitors such as etoposide [107]. The breakpoint in the AML1 gene on chromosome 21 may be identical to that in the t(8;21), but more often occurs approx. 60 kb downstream. The chromosome 3q26 locus is the site of three different genes that are involved in the t(3;21): from telomere to centromere, EAP, MDS1, and EVI 1. These genes are located within 200 kb of each other. In each of these translocations, the 5′-end of the AML1 (RUNX1) gene, including sequences that encode the runt-like domain, is fused to the 3′-end of one of these partner genes [107].
In the AML1–EAP fusion gene, the 3′-EAP sequence is very small and it is not fused to AML1 in reading frame. Thus, the AML1-EAP chimeric protein lacks transactivation activity and may exert its effect by inhibiting normal AML1 protein. In contrast, the AML1-MDS1 and AML1–EVI 1 fusion genes contain 3′-sequences of the MDS1 and EVI 1 genes, suggesting that their proteins possibly have unique functions, in addition to inhibiting normal AML1 protein. One potential explanation for the occurrence of these different translocations, not proven, is that the entire 3q26 region is transcribed as a result of the t(3;21), and different fusion genes result from alternative splicing of mRNA [108]. The normal EAP, MDS1, and EVI1 proteins are not normally expressed in hematopoietic cells, but are expressed as a result of the t(3;21).
30.2.2.8 AML with t(9;22)(q34;q11)
The existence of AML associated with t(9;22)(q34;q11) has been controversial in the past, in large part, because distinguishing AML from an initial presentation of CML in blast crisis (phase) can be difficult. The presence of Philadelphia chromosome and BCR/ABL fusion gene transcripts are not reliable criteria, as some cases of AML with these features have been reported [109], including cases in which BCR/ABL fusion transcripts were acquired in MDS. To complicate the issue, the karyotype in CML blast crisis is often very complex. Some authors believe that the only reliable way to distinguish these entities is by assessing therapeutic response or by testing maturing hematopoietic elements for the BCR/ABL, present in CML only. The t(9;22)(q34;q11) is rarely identified in apparently de novo cases of AML, in 1–2 % of cases [109, 110]. The t(9;22) involves the ABL1 gene at chromosome 9q34 and the BCR gene at chromosome 22q11. The translocation results in a BCR-ABL fusion transcript. Two different forms of the t(9;22) have been detected in AML that result in either a 190 kDa or 210 kDa fusion protein [110]. In both translocations, the brea kpoint in ABL1 occurs in the same general area, in the proximal portion of the gene. In the p190bcr-abl form, the breakpoint in BCR occurs in the first exon. In contrast, in the p210bcr-abl form the breakpoint occurs in the 5.8 kb major breakpoint region of BCR. The detection of the p190bcr-abl fo rm of the t(9;22) in a case of AML is evidence to support the diagnosis of de novo AML. NPM1 mutations have been identified in a subset of de novo AML associated with t(9;22) [109], but no ABL1 mutations, known to occur in a subset of untreated CML patients, have been reported [109].
30.2.2.9 AML with t(7;11)(p15;p15)
The t(7;11)(p15;p15) is a rare translocation in AML. Morphologically, most AMLs with the t(7;11) exhibit granulocytic maturation and are classified as FAB-M2; coexistent myelodysplastic features are common [111]. Most patients with t(7;11)-positive AML have been Japanese adults, suggesting a genetic predisposition for this type of AML [111].
The t(7;11) disrupts the HOXA9 gene at chromosome 7p15 and the NUP98 gene at 11p15, resulting in formation of the NUP98-HOXA9 and HOXA9-NUP98 fusion genes [105, 111]. The NUP98-HOXA9 fusion gene, located on the derivative chromosome 11, is thought to be involved in leukemogenesis, based on the predicted structure of the chimeric protein [105, 111]. The reciprocal HOXA9-NUP98 fusion gene is probably nonfunctional, and was not amplifiable by RT-PCR in a subset of cases tested [105]. The normal HOXA9 gene is a member of the homeobox gene family, and is involved in development and differentiation. The HOXA9 gene encodes a class I homeodomain protein that is expressed in hematopoietic cells and the kidney [105, 111]. The normal NUP98 gene is relatively small, with five exons, and encodes a component of the nuclear-pore complex that is 98 kDa.
30.2.2.10 Gene Mutations Associated with AML
FLT3 Gene
FMS-like receptor tyrosine kinase 3 ( FLT3 ) , also known as fetal liver kinase 2(FLK-2) and stem cell tyrosine kinase 1 (STK-1), belongs to a class III receptor tyrosine kinase family that also includes KIT, FMS, and platelet-derived growth factor receptor (PDGFR) [112]. The human FLT3 gene is located on chromosome 13q12 and contains 24 exons [112]. FLT3 has been demonstrated to exist in two forms: a membrane-bound protein of 158–160 kDa that is glycosylated at N-linked glycosylation sites in the extracellular domain and an unglycosylated protein of 130–143 kDa that is not membrane bound [113]. FLT3 is preferentially expressed by hematopoietic stem cells as well as in the brain, placenta, and liver [114]. The ligand of FLT3, which is expressed as a membrane-bound or soluble form by bone marrow stromal cells, stimulates the stem cells by itself or in cooperation with other cytokines [113, 115–117]. FLT3 is expressed on the surface of a high proportion of AML and stimulation of FLT3 enhances proliferation and reduces apoptosis [118].
Two unique forms of FLT3 gene mutation have been described. Initially, an internal tandem duplication (ITD) in the juxtamembrane domain–coding sequence of FLT3 was described (Fig. 30.12) [119]. Subsequently, a missense point mutation at the D835 residue and point mutations, deletions, and insertions in the codons surrounding D835 within a FLT3 kinase domain (KDM) were found [120, 121]. FLT3/ITD and FLT3/KDM occurs in 15–35 and 5–10 % of adults with AML, respectively, and are associated with a poor prognosis [122, 123]. In addition, extremely high levels of FLT3 transcripts have been demonstrated in a proportion of AML patients without FLT3 mutations, and are also associated with a poor prognosis [124]. Rarely, both types of mutations occur simultaneously in AML; this may be related to clonal progression [125].
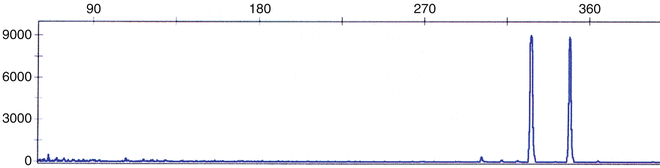
Fig. 30.12
Multiplex fluorescence-based PCR method used to detect internal tandem duplication (ITD) of FLT3. DNA is assessed by amplification of the juxtamembrane domain using primers from exons 11 and 12. The presence of second, larger peak signifies that FLT3/ITD is present.
FLT3 mutations lead to constitutive activation resulting in increased receptor signaling, which in turn is invovled in tumorigenesis. FLT3/ITD is constitutively phosphorylated on tyrosine residues and forms homodimers and, if the ITD-containing FLT3 is cotransfected with wild-type FLT3, a heterodimer with wild type FLT3 [122]. FLT3/ITD or FLT3/KDM result in autonomous proliferation of murine interleukin 3-dependent cell lines, such as 32D, and induce constitutive activation of downstream signaling molecules, such as signal transducer and activation of transcription 5 (STAT5), MAP kinase, SHC, AKT, and BAD [122, 126, 127]. 32D cells are known to differentiate into mature neutrophils in response to granulocyte colony-stimulating factor (G-CSF). However, both FLT3/ITD-expressing and FLT3/KDM-expressing 32D cells do not differentiate into mature neutrophils when they are treated with G-CSF [128]. These results indicate that FLT3 mutations are involved in cascades for autonomous cell proliferation and differentiation block. In addition, mutant FLT3 has antiapoptotic effects on leukemia cells [127].
It has been shown that FLT3/ITD induces an oligoclonal myeloproliferative disorder in mice, but not AML, suggesting that additional mutations impairing hematopoietic differentiation and/or proliferation may be necessary for AML development [129]. Transplantation of FLT3/ITD-transfected bone marrow cells into PML/RARα transgenic mice clearly shortens the latency period and increases penetrance for developing acute promyelocytic leukemia-like disease [130].
FLT3 mutations are mainly found in de novo AML, and are less frequent in AMLs that have developed from MDS or therapy-related AML [131, 132]. In low grade MDS, FLT3/ITD or FLT3/KDM occurs in approximately 3 % of cases. However, the frequency increases in advanced-stage MDS, and in up to 15 % in patients with AML that has developed from MDS [131, 132]. The frequency of FLT3 mutations in AML is associated with patient age. FLT3/ITD has been found in about 25 % of adult AML patients, but is higher (~30 %) in patients older than 55 years [133]. In contrast, FLT3/ITD has been found in approximately 10 % of pediatric AML patients [134, 135] and is rare in infant AML. FLT3 mutations are frequently found in AML with normal or intermediate-risk cytogenetic characteristics and with t(15;17), but are infrequent in patients with core binding factor translocations, such as t(8;21) and inv(16). Recently, it has been demonstrated that the NPM1 gene is mutated in more than 60 % of adult AML patients harboring FLT3 mutations [77–79].
FLT3/ITD is strongly associated with leukocytosis and an increased percentage of blast cells in the peripheral blood and BM of AML patients [131, 132]. The association of FLT3/KDM with leukocytosis is controversial. Only one study has demonstrated significant leukocytosis in adults who have AML with FLT3/KDM [121]. Several large-scale studies have shown the impact of FLT3 mutations on patient outcome. FLT3/ITD is a strong adverse predictive factor for overall survival, disease-free survival, and event-free survival within the intermediate-risk cytogenetic category. The clinical impact of FLT3/KDM on long-term outcome is controversial. Recent meta-analysis of four published studies including 1160 adult AML patients suggests an adverse effect of FLT3/KDMs on long term outcome [136].
RAS Gene
RAS is a GTP-dependent, 21 kDa protein that is localized at the inner side of the cell membrane. RAS is encoded by the RAS gene family, including KRAS located at 12p12.1, HRAS located at 11p15.5, and NRAS located at 1p13.2. RAS binds guanine nucleotides, displays intrinsic GTPase activity, and transduces signals from growth factor receptors to downstream effectors. The GTPase-mediated cycling between RAS bound to GTP (active conformation) versus GDP (inactive conformation) serves as a regulatory switch for signal transduction.
In AML, activating somatic mutations in the first two exons of RAS have been described, including codons 12, 13, and 61 of N-RAS, and sporadically in K-RAS [137]. These point mutations disrupt RAS function by diminishing GTPase activity. The reported frequency of N-RAS mutations in AML ranges from 10 to 27 % [137]. K-RAS mutations at the same codons can be found in approximately 5–10 % of pediatric and adult AML patients [138]. H-RAS mutations are rare in hematologic tumors [137]. In two studies, the presence of RAS mutations correlated with low blast counts in the BM and a better survival of patients in an age-adjusted analysis [139]. However, in other reports patients with RAS mutations showed a trend to a worse outcome compared to patients without tyrosine kinase or RAS mutations [138].
Patients with MDS and N-RAS mutations have a very high risk of progression to AML suggesting that these mutations might represent an important progression factor in MDS. On the other hand, analyses of RAS mutation status in patients at first diagnosis and at relapse revealed a marked instability of these mutations, with loss of mutations in some cases or new mutations in different codons. This may indicate that RAS mutation is not a leukemia-initiating event [140]. RAS has been shown to be an important mediator of G-CSF-induced and thrombopoietin-induced differentiation and to enhance the function of CEBPA in promoting myeloid differentiation [141].
KIT Gene
Activating mut ations of KIT, which are well-known in mast cell disorders and in gastrointestinal stromal tumors, also occur in AML. These mutations usually occur in one of two regions: the juxtamembrane region that functions as a negative regulatory region [142], and residues in the activation loop. KIT mutations are infrequent in AML in general, but are more common in patients with core binding factor AMLs, ranging from 2 to 20 % [143].
G-CSF Receptor
Approximately 20 % of patients with severe congenital neutropenia have G-CSF receptorpoint mutations. These mutations result in a truncation of the C-terminal region which is crucial for G-CSF signaling [144]. The mutated receptor interrupts signals required for normal myeloid cells. Patients with these mutations have an increased risk of progression to AML [144]. In 32D cells, the expression of a truncated G-CSF receptor resulted in increased proliferation without neutrophilic differentiation [145]. Transgenic mice expressing mutant G-CSF receptor (truncated at amino acid 715) develop a form of neutropenia in which myeloid progenitors react to G-CSF with hyperproliferation and neutrophilia [146]. Transgenic mice overexpressing a form of the receptor truncated at amino acid 718 or 731 have low neutrophil counts compared with wild-type controls and increased numbers of immature myeloid cells in the bone marrow [146, 147]. These effects are attributed to ligand-induced internalization of the receptor [148], reduced ability of the mutant receptor to activate STAT3, and reduced STAT5 inhibition by SOCS3 [149]. These features might be responsible for increased risk of AML in patients with severe congenital neutropenia.
IDH1 and IDH2
The isocitrate dehydrogenase (IDH) family of enzymes has three members: IDH1, IDH2, and IDH3. These enzymes catalyze the oxidative decarboxylation of isocitrate to produce CO2 and α-ketoglutarate (αKG). IDH1 and IDH2 are NADP+-dependent while IDH3 is NAD+-dependent. IDH1 contains a C-terminal peroxisomal localization sequence and is localized in both peroxisomes and cytosol; IDH2 and IDH3 are located in mitochondria [150]. A mutation in the IDH1 gene was first discovered in colorectal cancer and subsequently shown in brain gliomas. Later, mutations in IDH1 and IDH2 were identified in AML as well as MDS [151, 152]. In addition, IDH2 mutations have been identified in T-cell lymphomas, but to date, there have been no reports of cancer-associated mutations in IDH3.
In the initial report by Mardis and colleagues, IDH1 R132 mutation was detected in approximately 8 % of AML cases analyzed and was associated with normal cytogenetics [153]. Subsequently IDH2 mutations were detected in patients with AML. The initial study cited the frequency of IDH1 and IDH2 mutations in patients with AML as 7.7 and 15.4 %, respectively [153]. In subsequent studies, focused on a relatively homogenous group of adult patients with de novo AML with diploid cytogenetics, mutations in IDH1 and IDH2 were found in 14 and 19 % of cases, respectively [151, 152]. IDH1 and IDH2 mutations are not mutually exclusive as several patients harboring both an IDH1 and IDH2 mutation have been detected [152, 155]. Unbiased sequencing also has revealed the presence of 12 nonsynonymous recurring mutations (including IDH1) in more than one AML genome. The data implicate these alterations as important for leukemogenesis [153].
Recent studies across large patient cohorts have shown that IDH1 and IDH2 mutations are strongly associated with the presence of NPM1 mutation [153–156]. An analysis of cytogenetically diploid AML patients has shown that IDH mutations adversely affect prognosis [152]. Retrospective clinical trial data also have shown that IDH1 or IDH2 mutations are associated with a worse prognosis in younger AML patients with diploid cytogenetics and mutated NPM1 without FLT3-ITD [152, 155].
TP53 Gene
The TP53 tumor-suppressor gene, located at chromosome 17p13, is mutated in a wide variety of human cancers including AMLs. TP53 encodes for a nuclear phosphoprotein that binds to DNA and influences the expression of a variety of genes. p53 protein appears to be involved in cell proliferation, apoptosis, and DNA repair. Over 200 cases of AML have been assessed for TP53 abnormalities. Point mutations were identified in about 6 % of cases. TP53 gene mutations have been identified in all morphologic types of AML except acute promyelocytic leukemia. Presence of TP53 mutations correlates with older patient age, presence of myelodysplasia, and poorer prognosis [157].
30.2.2.11 Other Abnormalities in AML
STAT Activation
Consitutive activation (phosphorylation) of signal transducer and activator of transcription (STAT) 3 and STAT5 has been shown in cases of AML [126]. For full transcriptional act ivity, STAT proteins must be phosphorylated on serine residues, often by MAP kinases, and this also has been observed in AML blasts. Aberrant regulation of gene expression contributes to constitutive activation of STAT proteins. STAT3 and STAT5 can be activated by mutationally activated isoforms of KIT and FLT3 in AML blasts [126]. SOCS proteins are potent repressors of STAT activation, and their ability to repress transcription occurs through aberrant DNA-methylation of regulatory regions upstream of their transcriptional initiation site.
Constitutive activation of STAT3 and STAT5 appears to work as a signal integration site for several pathways on the way to leukemic transformation. The transforming ability of constitutively activated STAT5 protein on mouse bone marrow was demonstrated using retroviral transduction/transplantation studies. STAT3 or STAT5 are necessary for tyrosine kinase-mediated transformation of myeloid cells.
CXCR4 Expression
CXC chemokine receptor 4 (CXCR4 ) is one of a number of chemokine receptors defined by their ability to induce cell migration towards a chemotactic cytokine gradient (chemotaxis). CXCR4 has received much attention in the literature because it is the receptor for stromal derived factor (SDF-1α), also known as CXCL12, and the CXCR4-CXCL12 axis is essential for the migration of normal cells to the bone marrow microenvironment [158]. CXCR4-CXCL12 also appears to play a role in metastases of both hematopoietic and solid tumors [158, 159]. CXCR4 is expressed and functional in a subset of AML cases and expression correlates with poorer prognosis [159, 160]. In addition, CXCR4 has been reported to be expressed more frequently and at a higher level in AML cases associated with FLT3/ITD mutations [161]. Based on this last observation, a link between CXCR4 expression and FLT3/ITD mutation has been proposed. One group has demonstrated that CXCR4 expression is associated with a poorer prognosis in AML patients with normal cytogenetics and unmutated FLT3 [160].
Antigen-Receptor Gene Rearrangements in AML
Immunoglobulin heavy chain (IgH) and /or T-cell receptor (TCR) β-chain and γ-chain gene rearrangements have been detected in less than 5 % of cases of AML, using Southern blot analysis [162]. However, a study of AMLs using PCR-based methods found a substantially higher number of cases with monoclonal IgH gene rearrangements. T-cell receptor rearrangements are more common in AML cases with stem cell immunophenotypic features. Many AML cases with antigen-receptor gene rearrangement s are undifferentiated, often TdT-positive, and express one or more lymphoid-associated antigens, suggesting that these leukemias are arising from an early BM precursor cell with multilineage potential [162]. Patients with AML associated with antigen receptor gene rearrangements are reported to have a poorer prognosis than patients with AML without these gene rearrangements.
30.2.2.12 Therapy-Related AML
Patients with therapy-related acute myeloid leukemia (t-AML ) present either early, within 3 years of cessation of all chemotherapy, or late, 7 or more years after completion of therapy. Early t-AMLs often display balanced translocations, especially involving the MLL gene, whereas late t-AMLs often shown unbalanced rearrangements, cytogenetic abnormalities of chromosomes 5 and 7, and are often preceded by MDS [163]. Clinicopathologic studies with multivariate analysis have implicated topoisomerase II inhibitors, particularly the epipodophyllotoxins etoposide and teniposide, in the generation of balanced MLL translocations in the patients with early t-AML [164]. Data from the initial series of cases with t-AML related to topoisomerase II inhibitors suggested that the epipodophyllotoxins were more highly associated with t-AML than other topo II agents, and that dose and schedule of therapy were important.
Cytotoxic chemotherapeutic agents are associated with cases of late t-AML. Gene mutation studies using next-generation sequencing methods have shown that mutation profiles in t-AML differ greatly from de novo AML cases. In particular, TP53 mutations are much more common in t-AML.
30.2.2.13 AML in Patients with Down Syndrome
Children with Down syndrome (DS) have a 10-fold to 20-fold increased risk of developing acute megakaryoblastic leukemia when compared with their non-DS counterparts [165]. Recently, an important role for the hematopoietic transcription factor GATA1 has been demonstrated in DS patients with either transient myeloproliferative disorder (also known as transient abnormal myelopoiesis) or acute megakaryoblastic leukemia [166]. Expression of GATA1 is essential for normal maturation of erythroid cells and megakaryocytes [167]. In two studies of 41 DS patients with acute megakaryoblastic leukemia or transient myeloproliferative disorder, missense mutations in GATA1 were identified in over 90 % of cases; the mutations were not identified in subsequent remission samples and were determined to be acquired [168]. In contrast, GATA1 mutations were not seen in four cases of acute lymphoblastic leukemia in DS patients [168].
Genomic profiling studies have shown that cases of transient myeloproliferative disorder often have only trisomy 21 and GATA1 mutations. In contrast, in patients that progress to acute megaryoblastic leukemia a number of other genes are mutated including cohesin genes, genes that regulate epigenetics (e.g., EZH2), CTCF, JAK family genes, MPL, and RAS pathway genes [169].
30.2.2.14 Hypoxia and Hypoxia-Related Changes
Recently, hypoxia and hypoxia-related changes in the BM have attracted the attention of investigators analyzing AML and other malignancies. In normal BM, hypoxia is critical for development of hematopoietic stem cells [170]. A critical role in mediating the effect of hypoxia on hematopoietic stem cells is played by hypoxia-inducible factor 1 alpha (HIF1α) [171]. Decrease in oxygen tension in BM can lead to the selective outgrowth of AML clones with a survival advantage in the severely hypoxic microenvironment [172]. Data obtained from animal models indicate that AML progression within BM is associated with further decreases in overall oxygenation and expansion of hypoxic areas [172]. Hypoxia also affects the exposure of AML cells to systemic chemotherapy and the antitumor immune response [173]. The overexpression of HIF1α in primary AML and acute lymphoblastic leukemia BM samples correlates with poor chemotherapy outcomes [174, 175]. For these reasons, hypoxia-activated agents, novel cytotoxic drugs preferentially activated under low oxygenation conditions, may have an active role in the treatment of AML and other BM malignancies. Recent studies have demonstrated that HIF1α plays a role in cancer progression by activating transcriptional programs for maintaining the self-renewal and multipotent capacities of cancer stem cells in a hypoxic environment [176–178]. HIF1α was also shown to be required for stem cell functions in mouse lymphoma and human AML [179]. One of the new approaches in AML therapy is hypoxia-activated prodrugs such as TH-302. TH-302 is a 2-nitroimidazole hypoxia-activated prodrug of the cytotoxin bromo-isophosphoramide mustard. In vitro, TH-302 demonstrates activity against solid tumors, multiple myeloma cells, and AML cells under low oxygen tension [173, 180, 181]. In clinical trials, TH-302 alone and in combination with chemotherapy was very effective in solid tumors and hematologic malignancies including AML and multiple myeloma [182, 183].
30.3 Blastic Plasmacytoid Dendritic Cell Neoplasm
Blastic plasmacytoid dendritic cell neoplasm (BPDCN ) is a rare tumor that arises from precursors of plasmacytoid dendritic cells (also known as plasmacytoid monocytes or professional type 1 interferon producing cells). The age range of patients with BPDCN is quite broad, from young children to the elderly and males are more often affected. Patients with BPDCN usually present with skin disease and involvement of bone marrow. Most patients also present with low-level peripheral blood involvement and lymphadenopathy occurs in about 50 % of patients. Overall survival is poor, in the range of 1–2 years despite therapy [184, 185].
Conventional cytogenetic analysis commonly shows a complex karyotype but there are no consistent recurrent abnormalities. Deletions of a number of chromosomal loci have been reported with del(5q) most common. Rare cases reported have had T-cell receptor gene rearrangements, but in most cases of BPDCN the IgH and TCR genes are germ line.
Gene expression profiling of BPDCN has pointed to activation of the NF-kB pathway as a potential therapeutic target [186]. A number of gene mutations have been identified using whole exome or more targeted sequencing approaches including TET, IKZF3, HOXB9, UBE2G2, and ZEB2. A single case of a child with BPDCN associated with a CLTC-ALK fusion gene has been reported [187].
TET mutations are most common in BPDCN, in up to 40 % of cases [184]. TET is also mutated in cases of MDS and AML. TET normally converts 5-methyl-cytosine to 5-hydroxymethylcytosine and is expressed in early hematopoietic precursors. TET is involved in epigenetic regulation and hematopoiesis. TET mutations are usually heterozygous and result in loss of function, presumably allowing uncontrolled expansion of hematopoietic cells [187].
30.4 Acute Lymphoblastic Leukemia
Acute lymphoblastic leukemia (ALL) is an umbrella term for a group of neoplasms composed of immature lymphoid cells that are derived from the bone marrow. Previously, the FAB system classified these neoplasms on the basis of their morphological features as L1, L2, or L3. It is now recognized that the distinction between L1 and L2 has little biological and clinical meaning, and that L3 cases represent Burkitt lymphoma/leukemia [188].
Far more important in ALL are their immunophenotypic and molecular features. Cases of ALL can be divided into immature B-cell and T-cell neoplasms and each of these categories has a number of molecular subgroups. Approximately 80–90 % of ALL cases are of immature B-cell lineage, and include neoplasms once designated as non-B/non-T, null, and common ALL. The remaining 10–20 % of ALL cases are of T-cell lineage. Heritable syndromes predisposing to ALL and germ line mutations in known tumor-suppressor genes are uncommon, implying that most cases probably arise from somatic mutations in immature lymphoid cells.
Immature B-cell and T-cell neoplasms can present as either ALL or lymphoblastic lymphoma. At the molecular level leukemic and lymphomatous B-cell or T-cell neoplasms appear to be similar. For this reason, the current WHO classification designates these tumors as B or T lymphoblastic leukemia/lymphoma. However, as most studies of these tumors have focused on cases that presented as leukemia we use the terms B-ALL or T-ALL or lymphoblastic lymphoma (LBL).
30.4.1 B-Cell ALL
Based on antigen expression, B-ALLs can be further subclassifi ed into as many as five subgroups: (1) pre-pre-B-ALL, (2) early pre-B-ALL, (3) pre-B-ALL, (4) transitional B-ALL, or (5) mature B-ALL, although subdivision into fewer subgroups based on the expression of CD10, cytoplasmic IgM, and surface immunoglobulin is the usual practice. Nearly all B-ALLs carry monoclonal IgH gene rearrangements. In addition, 40–50 % of ALLs also contain Igk gene rearrangements, and 20–25 % contain Igλ gene rearrangements. The results suggest a hierarchy of Ig gene rearrangements in developing B lymphocytes: IgH rearrangement first followed by Igk and then Igλ.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


