Fig. 35.1
MRI sequences of common primary brain tumors. (a) Sagittal T1-weighted and gadolinium-enhanced MR image showing a medulloblastoma in the posterior fossa (arrow) with compression of the pons and medulla oblongata; (b) Axial T2-weighted MR image showing a space occupying ependymoma (hyperintense signaling, arrow) in the left paramedian posterior fossa resulting in displacement of the brain stem; (c) Sagital T1-weighted and gadolinium-enhanced MR image depicting a slightly and inhomogenously enhancing low grade astrocytoma in the thalamic and mesencephalic region (arrow); (d) Axial T1-weighted and gadolinium-enhanced MR image showing a centrally located Glioblastoma (arrow) in the region of basal ganglia resulting in displacement of the pyramidal tracts (brown fringe).
The management strategies of human brain tumors rely mostly on histopathological characteristics that are used to predict the clinical behavior and eventual patient prognosis of a diagnosed brain tumor. Features including degree of anaplasia, mitosis, infiltration, necrosis, and neovascularization define the corresponding tumor grade, which ranges from benign, WHO grade I, to the most malignant, WHO grade IV [9]. The advances in molecular diagnostics over the last 5 years however have led to rapid genetic profiling of many brain tumors, which allow us now to recognize remarkable differences in prognosis for tumors harboring similar histopathological patterns [10, 11]. Currently, the utilization of some molecular markers informs individualized treatment decisions for patients with the same histological tumor [10]. Future revisions of brain tumors classification systems need to implement molecular profiles in order to support the differential diagnosis and to be of value in predicting the efficacy of treatment options.
Despite concerted efforts in the field of neuro-oncology , brain tumors represent perhaps the most intimidating and difficult to treat type of cancer. Compared to other neoplasms, patient with malignant gliomas still face disproportionately high rates of morbidity and mortality [12] (Table 35.1). Fortunately, recent advances in molecular biology have provided a wealth of new information that can now be applied to improving patient treatment strategies and outcome. Modern medicine is on the threshold of achieving many tangible gains in overall patient survival based on the progress that has been made in several areas of experimental neuro-oncology.
Table 35.1
Clinical characteristics of common primary brain tumors
Tumor | Preferential location | Age | Gender | Annual incidence | WHO grade | Present treatment strategy | Prognosis |
|---|---|---|---|---|---|---|---|
Medulloblastoma [1, 424–427] | Cerebellar | Median: 8 y 93 % < 15 y | m: f ~ 1.6:1 | 0.6/100,000 (age 1–9) | Grade IV | – Maximal safe resection followed by C (<3 y) and RT (>3 y) – High-dose C and autologous stem cell transplantation with or without RT to be considered, especially for high-risk group – Re-resection at progression followed by C with or without RT | Pediatric: – 5-y survival • 42 % (infants < 1 y) • 72 % (1–9 y, including high- and average-risk (ar)) – 10-y survival for ar group: • 81 % Adults: – 5-y survival • 67 % WNT group correlates with good prognosis (>90 % long-term survivals) |
Intracranial ependymoma [1, 208, 428] | Infratentorial (60–70 %), supratentorial (30–40 %) | Median: 8 y | m: f ~ 1.1:1 | 0.3/100,000 | Grade II (~50 %) Grade III (~50 %) | – Maximal safe resection followed by RT – C for Grade III, non-resectable tumors, low EOR and at progression – Re-resection at progression followed by RT with or without C | Pediatric: – 5-y survival • 66–79 % – 10-y survival • 55–68 % |
Pilocytic astrocytoma [1, 247, 249, 429] | Cerebellar (in children > 50 %), optic pathway, hypothalamus | Median: 18 y 38 % < 15 y 75 % < 20 y | m: f ~ 1:1 | 0.9/100,000 (age 0–19) | Grade I (>90 %) | – Maximal safe resection – Post-OP: • Usually no additional therapy, depending on EOR • RT optional for non-resectable tumors and at progression • C optional for non-resectable tumors and at progression – Re-resection at progression | Pediatric: – 10-y survival Cerebellar: 100 % Overall: 97 % Adults: – 5-y survival Overall: 84 % |
Diffuse low-grade glioma (DLGG) – Astrocytoma (A) – Oligodendroglioma – (OD) – Oligoastrocytoma (OA) [107, 430-432] | Frontal (50 %), temporal (25 %) Insular (10 %) | Median: 45 y | m: f ~ 1.3:1 | 1/100,000 – A 20–50 % – OD 35 % – OA 10–45 % | Grade II | – Maximal safe resection – Post-OP: • C to be considered, especially for OD, non-resectable DLGG, low EOR and at Progression • RT for non-resectable DLGG and at Progression – Re-resection at progression, followed by C and/or RT | 5-y survival – >95 % After GTR – 78 % if Resection < 50 % 10-y survival – 90 % After GTR – 54 % if resection < 50 % OD and LOH 1p/19q tend to correlate with better outcome |
High-grade/anaplastic (A) glioma – AA – AOD – AOA [121, 206, 433–435] | Parietal, frontal, temporal | Median: 43 y | m: f ~ 1.4:1 | 0.6/100,000 | Grade III | – Maximal safe resection followed by RT or alkylating C (AA, AOD and AOA without LOH 1p/19q) – Maximum resection followed by C with or without RT (AOD and AOA with LOH 1p/19q) – Re-resection and C/RT at progression | Median survival – 4.6- 6.5 y (AOD, AOA) 5-y survival ~50–65 % (AOD, AOA) ~15–50 % (AA) 10-y survival ~20–45 % (AOD, AOA) ~15–30 % (AA) AOD, IDH1 mutations and LOH 1p/19q correlate with increased sensitivity to RT/C and better outcome |
Glioblastoma [433, 436, 437] | Temporal, frontal, parietal | Median: 57 y | m: f ~ 1.7:1 | 5/100,000 | Grade IV | – Maximal safe resection, followed by RT plus concurrent TMZ, followed by adjuvant TMZ (age < 65–70 years) – Maximal safe resection, followed by RT or TMZ with or without RT based on MGMT status (age > 65–70 years) – Re-resection and C/RT at progression to be considered | Median survival – 14.6 mt (overall) – 23.4 mt with methylated (met) MGMT promoter 5-y survival – 9.8 % (overall) – 13.8 % with met MGMT IDH1/2 mutations and met MGMT correlate with better outcome |
35.2 The Familial Central Nervous System Tumor Syndromes
Most CNS tumors occur in adults and arise spontaneously with no previous family history or genealogic pattern of inheritance. In some cases, especially in younger patients, they may present as a familial syndrome and transmitted most frequently in an autosomal dominant (AD) pattern of inheritance. Advances in the identification and characterization of gene mutations have permitted insight into the molecular basis of both familial and sporadic CNS tumors.
Many familial CNS cancer syndromes are associated with mutations in tumor suppressor genes, which mediate functions such as DNA repair and cell cycle arrest. Affected individuals typically inherit a germ-line mutation in one copy of the genetic locus and in the cases of non-AD mutations, are associated with somatic loss of the wild-type allele in order to promote the initiation of tumor growth. One important distinction of the familial syndromes is the predisposition of patients to develop multiple tumors in the CNS as observed in neurofibromatosis 1 (NF1), neurofibromatosis 2 (NF2), von Hippel–Lindau disease, tuberous sclerosis complex, Gorlin syndrome, Li–Fraumeni syndrome, Turcot syndrome, and Cowden syndrome.
35.2.1 Neurofibromatosis Type 1
NF1, also known as von Recklinghausen’s disease, is one of the most common inherited monogenetic syndromes predisposing individuals to the formation of CNS tumors, with an incidence of 1 in 3500 individuals and an equal predominance in both genders [13] (Fig. 35.2). NF1 is AD, with marked pleiotropy and variability in clinical expression. Approximately 50 % of newly diagnosed patients have no previous family history, suggesting the occurrence of de novo mutations, with a bias for the paternal germ-line. The disease is caused by loss-of-function mutations of the NF1 gene. The diagnosis of NF1 is made according to well defined clinical criteria (NIH criteria) [14], which are usually diagnostic by the age of 8 years [15]. Detecting mutations in the NF1 gene is difficult and complex given the large size of the gene and the lack of established mutation hotspots. Protocols combining PCR and high-performance liquid chromatography to detect germ-line mutations have been developed and have reported the identification of more than 95 % of individuals fulfilling clinical NF1 criteria [16].
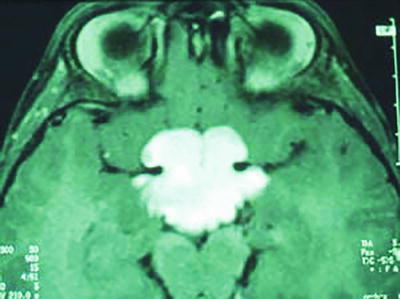
Fig. 35.2
T1-weighted and gadolinium-enhanced MRI sequence showing a Pilocytic Astrocytoma of the optic pathway in a patient with neurofibromatosis type 1.
The disease is characterized by the presence of multiple neurofibromas, pilocytic astrocytoma within the optic nerve pathways, malignant peripheral nerve sheath tumors (MPNST), and susceptibility to other astrocytomas. The optic pathway astrocytoma (OPA) represents the most common CNS tumor in NF1, affecting up to 20 % of all patients and usually arising in children under the age of six [17]. Remarkably, half of the individuals with OPA remain asymptomatic [17].
NF1 is inherited with high penetrance but the phenotypic expression is variable, suggesting a role for other disease-modifying genes and heterogeneous mutations of the NF1 gene [18–20]. The timing of NF1 gene inactivation or mutation is also assumed to interfere with disease manifestation [20]. The NF1 gene is localized to chromosome 17q in 1987 and mapped to the 17q11.2 locus 3 years later [21–23]. The NF1 gene encodes the protein Neurofibromin and spans at least 350 kilobases (kb) and includes over 60 exons. More than 1000 mutations resulting primarily in Neurofibromin truncations have been identified [24] and complete gene deletion is associated with a severe phenotype [25].
Neurofibromin functions as a tumor suppressor and is a cytoplasmic protein of that is highly expressed in neurons, Schwann cells, oligodendrocytes, astrocytes, leucocytes and the adrenal medulla, while its expression in other tissues is minimal [26]. Neurofibromin harbors a Guanosine Activating Protein (GAP)-related domain, conferring homology to other guanosine triphosphatase (GTP) activating proteins [27]. Ras is a GTP-binding oncoprotein, which promotes cell growth and differentiation. By accelerating the conversion of active GTP to its inactive, guanosine diphosphatase (GDP)-bound form, Neurofibromin acts as a negative regulator of Ras. Defects in Neurofibromin are thought to increase cell growth and facilitate tumor formation due to loss of Ras regulation. In addition, Neurofibromin regulates other proteins including adenylate cyclase in astrocytes, and potentially plays a role in neural stem cell proliferation, survival, and astroglial differentiation [28, 29].
While most of the neoplasms encountered in NF1 patients are not considered malignant, adults with NF 1 have a 50–100-fold increased risk of high-grade glioma formation [30, 31] and a 20 % lifetime risk to develop a malignant peripheral nerve sheet tumor (MPNST) [32, 33] which are assumed to arise from preexisting neurofibromas [32]. To date, only two genetics alterations have been identified as progression pathways. Mutation in the TP53 loci and genetic alterations in INK4A have been frequently identified in malignant but not benign forms of neurofibromas [30, 34].
Numerous animal models have been employed to explore the molecular basis of NF1 pathogenesis. NF1 heterozygous mice with selective inactivation of NF1 in Schwann cells develop tumors with histologic and molecular features of human neurofibromas [35]. The generation of astrocyte-specific NF1 knockout mouse yielded similar findings, with low-grade optic astrocytomas developing only when Neurofibromin expression was ablated in astrocytes of NF1 heterozygous mice [36].
Several therapeutic approaches based on blocking the dysfunctional neurofibromin pathway have been attempted in animal studies [37]. However, most clinical studies have not extended beyond Phase II trials [38]. The mTOR pathway is regarded as a key mediator of optic pathway glioma development. Currently, an ongoing Phase II study is investigating the growth inhibition of NF1 related low-grade gliomas using the mTOR-inhibitor everolimus [39].
35.2.2 Neurofibromatosis Type 2
NF2 is approximately tenfold less common than NF1 and has distinct phenotypic and genetic features. It is an autosomal dominant syndrome that occurs at an incidence of 1 in 25,000–40,000, with a penetrance close to 100 % by the age of 60 [40]. Half of the individuals inherit a germ-line mutation from a parent or acquire a de novo mutation at the stage of embryogenesis [41]. Bilateral vestibular schwannomas are pathognomonic of the disorder, which is also characterized by schwannomas of other nerves (cranial, spinal or peripheral) as well as intracranial and intraspinal meningioma and ependymomas. NF2 is characterized by a variable age at onset, with a majority of patients developing signs in the second to third decade of life, and thus, NF2 schwannomas and meningiomas occur at an earlier age compared to their sporadic counterparts [41]. NF2 is clinically diagnosed according to the 2005 Manchester criteria [42].
The NF2 gene was mapped to chromosome 22 in 1987 and further localized on locus 22q12.2 [43]. The gene responsible for this disorder was cloned in 1993 by two groups by positional cloning and was notably also found to be mutated or deleted in the majority of sporadic meningiomas and schwannomas [44, 45]. The gene spans 17 exons distributed approximately over 110 kb, and it encodes for a protein of 595 amino acids called merlin (for moesin, ezrin, radixin-like) due to its homology to three proteins of the ezrin, radixin, moesin (ERM) family [45]. Merlin, alternatively named Schwannomin, is expressed in many normal human tissues, including the brain, and functions as a tumor suppressor [46]. Tumors resulting from dysfunction or loss of Merlin activity are mostly benign [47].
Several studies have shown that Merlin controls cell proliferation and cell motility by regulating cytoskeletal organization, but the pathways involved in its mediation of growth suppression are poorly understood [46, 48]. Merlin interacts with F-actin and negatively regulates cell motility through cytoskeletal reorganization. Moreover, it may act with CD44 (a hyaluronic acid receptor), β1 integrin (a transmembrane glycoprotein) and paxillin (a cytoskeletal adaptor protein) to regulate cell motility [48]. Merlin deficiency has also been linked to abnormal activation and overexpression of receptor tyrosine kinases such as EGFR family receptors [49].
A number of NF2 mouse models have been developed. Homozygous mutation of NF2 results in embryonic lethality, indicating that its function is essential at early stages of mouse development [50]. Heterozygous NF2 mutant mice develop many neoplasms but do not manifest the classical tumors linked to NF2. However, as observed in NF1 murine models, the model of conditioned Schwann cells for knockout mice show many similarities with NF2 patients [51].
Merlin dysfunction or loss affects various signaling pathways, therefore several therapeutic targets have been proposed. As schwannomas represent the most common symptomatic and limiting tumor in NF2 patients, clinical trials are primarily directed against schwannoma formation [52]. A recent Phase II study in NF2 patients with progressive vestibular schwannomas observed some decrease in tumor volume and improvement in hearing in response to the EGRF inhibitor Lapatinib [53].
35.2.3 von Hippel–Lindau Syndrome
von Hippel–Lindau (VHL) is a rare inherited disease with an AD pattern of transmission and was first described in the early 1900s by Eugen von Hippel and Arvid Lindau [54, 55]. It is characterized by the development of benign hamartomatous tumors in the CNS and adrenal glands including hemangioblastoma and retina angioma, pheochromocytoma, pancreatic neuroendocrine tumors and inner ear tumors as well as malignant tumors of the kidney such as renal cell carcinoma [56]. Its incidence is estimated at 1 in 36,000–45,000 births per year [57, 58]. Up to 20 % of VHL patients are estimated to derive from de novo mutations [59]. The disease shows variable expressivity with almost 95 % penetrance at the age of 60 years [60]. A combination of characteristic clinical features, with or without a positive family history, permits the establishment of a clinical diagnosis. The most frequent lesion observed is hemangioblastoma, a mesenchymal solid and cystic tumor mainly localized in the brain (mostly located in the cerebellum), which occurs in 60 to 80 % of all VHL patients [61]. Depending on specific mutation in the gene locus, VHL is classified as type 1 or type 2, according to the absence or presence of a pheochromocytoma [56].
The gene responsible for this disease is the VHL tumor-suppressor gene, located on chromosome 3p25-26 [57]. Under normoxic conditions, the gene product targets hypoxia-inducible factors (HIF) for polyubiquitination and proteosomal degradation. Under hypoxic conditions, the accumulation of HIF will induce the transcription of hypoxia-regulated genes such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF) and transforming growth factor (TGF) -α. This explains the high vascularization of VHL-related tumors. Loss of the VHL gene can also result in aberrant expression of genes that control cell cycle progression and invasion [62, 63]. It has been shown in renal carcinoma cells that loss of the VHL gene results in the accumulation of hepatocyte growth factor (HGF), which interacts with the β-catenin pathway [64]. Loss of VHL protein activity has also been recently correlated to the activation of kinases required for Ras-induced cell transformation, thus representing a possible target for molecular ) inhibition [65].
Hemangioblastoma is a mesenchymal-derived hamartomatous tumor and patients with VHL-syndrome are not typically linked with glioma manifestation. However, there is some evidence that VHL expression interferes also with neuroepithelial tumor formation via suppression of HIF-1α/VEGF, which is coupled with inhibition of glioma proliferation and invasion [66].
35.2.4 Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is a multisystem autosomal dominant disorder, affecting 1 in 6000–10,000 individuals [67]. It was first described by Bourneville in 1880 and is characterized by CNS tumors including cortical hamartomas named tubers, subcortical glioneuronal hamartomas, subependymal glial nodules, and subependymal giant cell astrocytomas. TSC patients often suffer from debilitating neurologic disorders, including epilepsy, mental retardation, and autism. Essentially, any organ system can be involved and common features of TSC include dermatologic manifestations such as cutaneous angiofibromas, subungual fibromas, cardiac rhabdomyomas, renal angiomyolipomas, intestinal and visceral cysts, and pulmonary lymphoangiomyomatosis,
The diagnostic criteria consists of a set of major and minor features which appear at distinct points in development, but most patients have manifestations of the disease before 10 years of age [68]. Neurologic disabilities seem to be intimately related to the presence of cortical tubers, which occur in approximately 70 % of patients [69].
The penetrance of the disease is high but the clinical manifestations are considerably variable [70]. There is a high rate of new mutations, accounting for up to 85 % of cases [71]. TSC results from the mutation of one of two genes, TSC1 on chromosomes 9q34 and TSC2 located on 16p13.3. Clinical and pathological features caused by mutations in these two genes are indistinguishable, but the prognosis seems to be more severe when the disease is associated with a TSC2 mutation [72]. The TSC1 gene encodes a transcript of 8.6 kb called Hamartin, containing 23 exons, whereas the TSC2 gene encodes a transcript of 5.5 kb called Tuberin, containing 41 exons. Mutations in these genes can be identified in 60–80 % of TSC patients, with a wide spectrum of specific genetic alterations [73]. Hamartin and Tuberin function as a regulatory complex inhibiting the small GTPase RHEB and consequently limiting the activity of the mammalian target of rapamycin complex 1 (mTORC1), which is an intracellular regulator of cell growth and metabolism [74]. In a recent Phase I–II open-label study of patients receiving the mTOR-inhibitor everolimus, a clinically significant reduction in tumor volume of TSC related subependymal giant-cell astrocytoma was shown [75, 76].
35.2.5 Gorlin Syndrome
Gorlin syndrome is also known as nevoid basal cell carcinoma syndrome (NBCCS) or basal cell nevus syndrome (BCNS) and was first described by Goltz and Gorlin in the 1960s. It is a rare AD disorder occurring in 1 of 57,000 live births and characterized by basal cell-carcinoma, skeletal anomalies and a 2–5 % incidence of childhood medulloblastomas (mostly SHH medulloblastomas) [11, 77]. The penetrance is very high but the clinical expression is variable, with up to 30 % of patients presenting with de novo mutations [78]. The clinical criteria are based on the association of multiple basal cell carcinomas and odontogenic keratocysts [79]. Other manifestations include the presence of palm or sole pits, intracranial calcifications in particular along the falx and macrocephaly [77].
The disease results from a mutation of the human homologue of the Drosophila melanogaster gene PTCH located on 9q22.3 [80]. It contains 23 exons and encodes for a transmembrane protein (Ptch), which is a receptor and negative regulator for the secreted Hedgehog family proteins such as Sonic Hedgehog (Shh).
35.2.6 Li–Fraumeni Syndrome and TP53 Germ-Line Mutation s
Li–Fraumeni syndrome (LFS) is an AD disorder predisposing patients to an early onset of a wide spectrum of neoplasms. The syndrome was defined by Li and Fraumeni after they noticed a high incidence of cancer among family members of children with rhabdomyosarcoma [81]. Often described as a very rare syndrome with only several hundred identified individuals, its effective incidence is probably underestimated [82], in part also due to the existence of different classification schemes [83]. The most frequent tumors observed are breast cancer, sarcomas, osteosarcomas, brain tumors, acute leukemia and adrenocortical carcinomas.
The major genetic abnormality underlying this syndrome is a germ-line mutation of TP53, which has been identified in 70 % of LFS cases and in 22–44 % of families with the LF variant form [84]. Importantly, 50 % of the families with a TP53 germ-line mutation meet the diagnostic criteria of LFS. In the remaining cases of LFS without TP53 mutation, a germ-line mutation of the hCHK2 gene involved in G2 checkpoint control has been identified [84]. TP53 germ-line mutations occur most frequently in exons 5–8, with major hotspots on codons 245 and 248. In the 143 LFS families reported, the most frequently identified mutations are point mutations (85.3 % of cases). Interestingly, low grade and anaplastic astrocytomas of LFS patients harbor somatic IDH1 mutations at much higher rates compared to sporadic gliomas [85].
35.2.7 Turcot Syndrome
Turcot syndrome is an AD disorder characterized by the occurrence of adenomatous colorectal polyps or colon carcinomas and malignant brain tumors, mainly anaplastic astrocytomas, as well as glioblastomas and medulloblastomas (95 %) [86]. Turcot described the first cases in 1959, where two siblings developed malignant CNS tumors and numerous adenomatous colorectal polyps [87].
There are two major subgroups of Turcot syndrome. The type 1 pattern is characterized by an association of glioblastomas and hereditary non-polyposis colorectal carcinomas (HNPCC). This subgroup results from a germ-line mutation in DNA mismatch repair genes such as hPMS1 (ch 2q32), hPMS2 (ch 7p22), hMSH2 (ch 2p16), or hMLH1 (ch 3p21) [86]. These deficiencies lead to DNA replication errors and microsatellite instability, which is rare in brain tumors in the absence of Turcot syndrome [88]. In this subgroup, the mean age for occurrence of glioblastoma is lower than in the general population (18 years old versus 40–70 years old). The type 2 subgroup is characterized by association of WNT subgroup medulloblastomas with familial adenomatous polyposis (FAP). These patients tend to have a germ-line mutation in the adenomatous polyposis coli (APC) gene that lies on chromosome 5q21 [86]. In the type 2 Turcot syndrome subgroup, the median age of occurrence of medulloblastoma is 15 years, compared to 7 years of age in the general population.
35.2.8 Cowden Syndrome and Lhermitte–Duclos Disease
Cowden syndrome (CS) is a rare AD condition associated with multiple benign hamartomatous lesions mostly involving the skin and mucosa, but various organ systems including the cerebellum may be affected [89]. CS predisposes patients to malignant tumors, especially of the breast, thyroid, intestine, and endometrium [89]. The penetrance of CS is high and approaches over 90 % by the age of 20 years [89]. The syndrome was first documented in 1963 by Lloyd and Dennis and named after the first patient [90]. In 1991, more than 70 years after its first description [91], it was recognized that dysplastic cerebellar gangliocytoma or Lhermitte–Duclos disease (LDD) is often a manifestation of CS [92]. The prevalence of CS is estimated at 1:200,000, but the frequency is likely underestimated given that many non-symptomatic mucocutaneous manifestations related to this disease are also common in the general population and thus may not lead to extensive diagnosis [89]. Approximately half of the CS cases are recorded as inherited and half occur spontaneously [93]. The most common germ-line mutation) involves the tumor suppressor gene PTEN, located at the chromosome 10q23.3 [94, 95]. Early clinical data including nearly 50 CS families suggest that 85 % of patients with CS harbor a PTEN mutation [96, 97] However this may not be universally adopted given that application of risk assessment criteria from the National Comprehensive Cancer Network (NCCN) results in many patients diagnosed as CS despite the absence of PTEN mutations [98]. Germ-line mutations downstream of PTEN signaling were found in the PI3K/AKT pathway in up to 10 % of CS patients lacking PTEN gene alterations [99]. Not all patients with LDD show clinical manifestation of CS [100] and dysfunction or inactivation of PTEN is found in 67–83 % of LDD lesions [100, 101]. Selective inactivation of PTEN in neural cells of mice results in impaired migration of granular cell precursors and formation of cerebellar tumors histopathologically resembling LDD [102, 103]. Inactivation of PTEN is linked with increased levels of PI3K/AKT, which promotes cell growth and proliferation [102, 103]. Activated AKT regulates various pathways involved in protein synthesis and cell growth, including the mTOR pathway, suggesting that mTOR inhibitors may be effective in reversing cellular hypertrophy and tumor growth [101].
35.3 Cytogenetics and Molecular Genetics of Neuroepithelial Human Brain Tumors
35.3.1 Glioma (Astrocytoma and Oligodendrogliom a)
Gliomas comprise tumors of astrocytic or oligodendrocytic histopathological appearance. Whether these tumors have the same origin of tumor-initiating cells from neural progenitors or arise from dedifferentiated astrodendrocytes/oligodendrocytes is a matter of ongoing debate [104, 105]. The WHO distinguishes between gliomas with more circumscribed growth (WHO Grade I) and gliomas with diffuse infiltration (WHO Grade II: low grade, WHO Grade III–IV: high grade) [9]. Diffuse low-grade gliomas are considered a precancerous disease, as over time they progress to anaplastic, malignant tumors in most instances [106].
Gliomas demonstrate the full spectrum of cytogenetic and molecular abnormalities: From numerical and structural chromosomal alterations, to gene amplifications and overexpression, deletions and small-scale mutations up to and including epigenetic deregulations.
35.3.2 Chromosomal Alterations
Chromosomal number alterations occur with increased frequencies in high grade and adult gliomas, and chromosomal losses are observed more frequently than gains [107]. One of most common numerical autosomal changes seen in high-grade astrocytomas includes gain of chromosome 7, and loss of chromosome 10 [107–109]. Gain of chromosome 7 is seen in up to 83 % of adult high-grade astrocytomas, whereas chromosome 10 is reported to be lost in up to 86 % [109, 110]. In some studies, a combined gain of chromosome 7 accompanied by loss of chromosome 10 (7+/10–) occurs in over 80 % of GBM samples [108, 111].
The occurrence of trisomy 7 and monosomy 10 together has often been correlated with short-term survival, although currently available data does not allow a distinct conclusion [112]. Gains of chromosome 7 are also among the most common autosomal changes in low-grade gliomas (LGG), seen in 57 % of cases [110] and loss of chromosome 10 may be correlated with shorter overall survival [113]. Chromosomal alterations commonly observed in secondary GBM include 19q loss (see Box 35.1) [114]. An additional numerical chromosomal abnormality is the presence of double minute chromosomes, seen in approximately 20 % of GBM specimens, which can further confer oncogene amplification and drug resistance [115].
Box 35.1: Primary Versus Secondary GBM
According to current evidence, there are two different routes of glioblastoma tumorigenesis, both with different genetic alterations but ultimately undistinguishable histology [114]. GBM mostly occurs as a rapidly growing de novo tumor from a glial progenitor or dedifferentiated cell and affecting patients with a mean age around 60 years. This Primary GBM constitutes 90 % of the cases, where no evidence of a less malignant precursor lesion can be detected. Secondary GBMs (10 %) affect typically younger patients around a mean age of 45 years. These tumors progress from a preexisting diffuse low-grade or anaplastic astrocytoma, are more often located frontally, and the overall survival after diagnosis is significantly longer. Histologically, Secondary GBMs show less necrosis and frequent regions of oligodendroglioma-like appearance when compared to the Primary form. From a molecular basis, the Primary tumors often show loss of chromosome 10, EGFR amplification and PTEN mutation, whereas in Secondary GBM, TP53 mutations, loss of chromosome 19q and PDGFRA alterations are more frequent. Classification of GBM origin has been significantly improved by the recent identification of mutations in the enzyme gene IDH1/2 (isocitrate dehydrogenase, involved in the citric acid pathway), which are predominantly found in Secondary (80 %) but only rarely (<5 %) in Primary GBMs [132–134]. Consequently, IDH1/2 mutations are now commonly accepted as molecular markers of Secondary GBM.
Over 90 % of secondary GBM diagnosed through IDH1/2 mutations belong to the proneural subgroup, which indicates that this tumor subgroup is homogeneous compared to the primary form. IDH1/2 mutations occur early during progression from low-grade to high-grade glioma given they are detected in over 80 % of grade II and III astrocytoma and oligodendroglial tumors [134, 141]. It is possible IDH1/2 mutations precede 1p/19q loss in oligodendrogliomas and TP53 mutation in low-grade astrocytomas [242]. Finally, there is evidence that primary and secondary GBMs derive from different glial progenitor cells. In support of this, CD133 positive cancer stem cells could only be cultured from Primary GBMs, whereas samples from secondary GBMs did not have CD133 positive stem cells within [243].
Oligodendroglioma (OD ) and oligoastrocytomas (OA) constitute a subtype of glioma for which the specific loss of portions of 1p and 19q (loss of heterozygosity, LOH 1p/19q) is associated with important prognostic implications and is used as a marker to support the histological diagnosis of OD [116, 117]. Approximately 50–80 % of ODs have allelic loss of chromosomes 1p and 19q, whereas OA (mixed oligodendrogliomas, arrangement of cells presenting astrocytic or oligodendroglial differentiation) show this co-deletion 40 % of the time [118–120]. LOH 1p/19q is frequently observed in WHO grade II oligodendric tumors compared to those of grade III, though conflicting data exists in this regard [118–120]. Notably, oligodendric gliomas of the frontal lobe show higher incidence of 1p/19q co-deletion than those of non-frontal origin [119]. Low-grade OD (WHO II) treated by surgical resection and nitrosourea-based chemotherapy without radiotherapy show favorable long-term outcome (10-year overall survival rates over 90 %) irrespective of LOH 1p/19q status [121]. Anaplastic pure and mixed oligodendrogliomas (WHO III) identified with 1p/19q co-deletion are generally associated with marked higher overall survival and chemosensitivity in comparison to non-co-deleted WHO III oligodendrogliomas, where the addition of chemotherapy to radiotherapy did not prove beneficial over radiotherapy alone [120, 122].
35.3.3 Gene Amplification and Overexpression
Gene amplification has been described for several target genes in astrocytic tumors, and appears to be more common in high-grade lesions than low-grade astrocytomas [109, 123]. Genes continuously found to be amplified in high grade gliomas include EGFR, MET, PDGFRA, MDM4, MDM2, CCND2, PIK3CA, MYC, CDK4, and CDK6 [124]. Amplification and overexpression of EGFR occurs frequently and predominantly in primary GBMs (60 % of the samples), but rarely in the secondary form [114, 125]. In up to 30 % of the cases this gene has undergone loss of exon 2–7, resulting in the production of a constitutively active, truncated EGFR receptor (EGFRvIII) [109, 126, 127]. At this time, Phase II and III vaccination trials using rindopepimut to stimulate immune response against the EGFRvIII peptide in GBM patients are ongoing [128, 129]
35.3.4 Gene Mutations and Deletions
Tumor suppressor gene silencing events in astrocytomas may be the result of gene deletions, mutations, or promoter methylation events. Primary GBMs are characterized by CDKN2A deletion (31-50 %), and PTEN mutation (32 %) [109, 130, 131]. While CDKN2A deletions occur in primary GBM, TP53 mutations appear more common in secondary GBM [109, 114]. In the progression of low-grade tumors to secondary GBM, TP53 mutations are the most common and early-detected genetic abnormality—seen in about 2/3 of precursor low-grade astrocytomas and secondary GBMs derived from them. TP53 mutations occur in primary GBM but at a lower frequency (35 %) compared to secondary GBM [108, 109, 130]. Additionally, in secondary GBM, 60 % of the TP53 mutations are clustered in two hot spot codons (amino acids 248 and 273), whereas in the primary form, TP53 mutations appear more equally distributed within exon 5–8 [108].
Mutations in the gene encoding for NADPH-dependent isocitrate dehydrogenase (IDH1) were identified from a large-scale genomic sequencing analysis of astrocytoma [131]. Since the initial description, multiple studies have shown that IDH1 mutations are common in low-grade diffuse astrocytomas, anaplastic astrocytomas and tumors of oligodendroglial origin [132–134]. Notably, there is evidence that IDH1 mutations precede TP53 mutations, suggesting an important role in tumor initiation [114]. A proportion of low-grade astrocytomas that lack IDH1 mutations (e.g., pilocytic astrocytoma, and ependymoma) contain mutations of the related IDH2 gene [134]. As IDH1 mutations are very common in secondary (>80 %) but markedly rare in primary GBM (<5 %), a consensus is that IDH1 mutation can serve as a molecular marker of secondary GBM, as this form is neither clinically nor histologically distinguishable from the primary form [114]. IDH1 status is important to clinicians as patients with tumors harboring IDH1 mutations have a survival benefit [135–139]. IDH mutations also seem to correlate with both TP53 mutational status and 1p/19q deletions, both of which predict survival [137, 140, 141].
35.3.5 Epigenetic Alterations
In addition to aberrant changes in the genome, epigenetic alterations have emerged as important factors in gliomagenesis [142]. Potentially reversible and more amenable to treatment than numerical or structural changes in chromosomes, studies of epigenetic deregulations is a promising strategy in glioma therapy. GBM cells showing high activity level of the DNA repair enzyme MGMT (O6-methylguanine-DNA methyltransferase) are more resistant to therapies including alkylating agent such as temozolomide (TMZ) [143]. About 40 % of GBM have a methylated MGMT promoter status resulting in epigenetic silencing of the MGMT gene and thus higher sensitivity to TMZ and distinctive longer survival with actual standard therapy [143, 144]. Hence, MGMT promoter methylation is a strong prognostic and predictive biomarker.
The increased awareness of the role of metabolic enzymes in astrocytoma pathogenesis has brought about a resurgence of interest in the Warburg Effect [145]. The Warburg effect first described in 1956 by biochemist Otto Warburg promotes the observation that tumors predominantly use a high rate of aerobic glycolysis to meet metabolic demand [146]. Recently, isoforms of both pyruvate kinase (PK) M1/M2 and hexokinase (HK1/HK2) have shown differential expression in glioblastoma, in which PKM2 and HK2 predominate, promoting glycolysis [147, 148].
35.3.6 Pediatric Versus Adult Gliomas
The genetic changes that are found in pediatric astrocytomas are distinct from those in the adult variant of these tumors. No consistent karyotypic abnormalities have been identified in pediatric low-grade astrocytomas [149, 150]. Even in high-grade gliomas, the number of chromosomal aberrations is generally lower from their adult counterparts with up to 15 % lacking detectably number irregularities [107]. Relatively frequent compared to adult GBM, some pediatric GBMs have gain of chromosome 1q (up to 30 %) and loss of 16p (up to 24 %), while numerical aberrations of chromosome 7 and 10 are relatively rare (<30 %) [151, 152]. Cytogenetic changes recurrently observed in pediatric anaplastic astrocytomas and GBM include gains of 1q, 5q, and loss of 6q, 9q, 10q, 12q, 13q, and 22q [152, 153]. In contrast to adult tumors, amplification of the EGFR gene is not common in pediatric astrocytomas and PDGF-driven signaling is preferentially activated [151, 154]. Notably, this feature is also prevalent in secondary GBM [155]. Similarly, pediatric astrocytomas rarely exhibit loss or mutation of PTEN. Aberrant activation of the BRAF proto-oncogene (7q34) by copy number alteration or formation of an abnormal fusion protein is an important marker of pilocytic astrocytoma but not diffuse infiltrating pediatric astrocytomas (see Box 35.2) [156–158]. Aberrant BRAF activation leads to increased signaling through the MAP kinase/ERK pathways, which results in high levels of mTOR activation, and ultimately to increased cell growth [159, 160].
Box 35.2: Pilocytic Astrocytoma
Pilocytic Astrocytoma (PA) is a non-infiltrative, slow growing, and often cystic tumor typically classified as WHO grade I. It is one of the most common CNS neoplasms in childhood accounting for ~20 % of all pediatric brain tumors [1]. Almost three-fourths of the patients are younger than 20 years [1]. Approximately half of PAs are located in the cerebellum, but they can occur throughout the neuraxis such as the optic pathway, hypothalamus, and spinal cord (mostly in pediatric patients) as well as in the cerebral hemispheres (mostly in young adults) [244]. Optic nerve PAs are often associated with NF1. Provided that the tumor is accessible and extensive surgical resection is performed, most patients show a favorable outcome with a 10-year survival rate of over 95 % [244–246]. Until recently, the genetic mechanisms underlying this disease were not extensively investigated. Early genomic analysis of tumor samples showed often balanced karyotypes, whereas whole chromosomal gains occurred in about 30 %, frequently affecting chromosomes 5 and 7 [247]. Activating alterations within the MAPK-signaling pathway are considered a hallmark aberration in the pathogenesis of PA, affecting 80–100 % of analyzed tumor samples [248, 249]. Consequently it is hypothesized that PA represents a single-pathway disease [249]. The most frequent genetic event leading to MAPK activation is BRAF-KIAAI549 (B-K) fusion, which produces a fusion protein that lacks the regulatory domain of BRAF [249, 250]. In addition, various MAPK pathway-activating fusions ) typically involving BRAF are described [249]. The somatic mutation rate is very low (<0.1/Mb) and BRAF, FGFR1, K/-NRAS, PIK3CA, CDKN2A, and NF1 are the main genes found to be mutated or rearranged [245, 249, 251]. Similar to other tumor types, the copy number changes and mutation rates positively correlate with patient age and potentially with an aggressive clinical course [245, 247, 249]. The distribution of oncogenic hits within the MAPK pathway may vary depending on tumor location: NF1 mutations are more often detected in optic PA, while PAs in other locations are dominated by BRAF activation [248]. BRAF fusions are more common in cerebellar PAs and mutations are typically seen in supratentorial PAs [248]. In adult PAs, BRAF V600E mutations appear to be very infrequent compared to the pediatric counterpart [245]. As B-K fusions are particularly rare in diffuse astrocytomas (~2 %), the presence of this event can be considered an adjuvant diagnostic tool for PA [245].
35.3.7 Core Signaling Pathways and GBM Subgroups
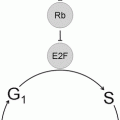
The understanding of the molecular basis of astrocytoma has benefited from large-scale genomic profiling approaches from networks such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) [109, 130, 131, 161] GBM was the first cancer to be systematically analyzed by TCGA, highlighting the role of oncogenes such as ERBB2 and PDGFRA and suppressor genes such as NF1 and TP53 as well as reinforcing core defects in three signaling pathways involved in the pathogenesis: (1) retinoblastoma (RB)-signaling (79 %), (2) p53 signaling (90 %), and (3) PIK(3)K/RAS signaling (88 %) (Fig. 35.3). Large-scale genomic consortia have allowed the stratification of astrocytomas based on genetic profiles giving valuable clinical information. The first subgrouping analysis on high-grade astrocytomas was reported by Philips et al. differentiating three distinct GBM clusters: proneural, proliferative, and mesenchymal, with the proneural subgroup predicting better survival [162]. A recent TCGA based study classified GBM into four molecular subgroups: classical, proneural, neural, and mesenchymal [155]. Genetic alterations in EGFR, PDGFRA/IDH1 and NF1 characterize the classical, proneural, and mesenchymal subgroups respectively [155]. Classical GBMs are further delineated by amplification of chromosome 7 paired with loss of chromosome 10, while often lacking TP53 mutations [155]. No distinctive genetic alteration is known that distinguishes the Neural from the other subtypes [124]. It still has to be elucidated to what extent the subgroup defining molecular profile may affect clinical outcome and sensitivity to specific therapeutic agents. DNA methylation profiling of 272 TCGA glioblastomas revealed a subset of tumors with a hypermethylation phenotype that was assigned to the proneural subgroup, further correlated with IDH1 mutations and in extended analysis also often found in low-grade gliomas [109, 163]. This glioma phenotype is an expression subtype and termed G-CIMP (CpG island methylator phenotype). G-CIMP positive gliomas affect younger patients, are probably more common in secondary GBM, and are associated with improved outcome [163]. In anaplastic oligodendroglioma the G-CIMP phenotype also consistently exhibit IDH1-mutations, MGMT promoter methylation, and LOH 1p/19q and is used as a predictor for better survival [164].
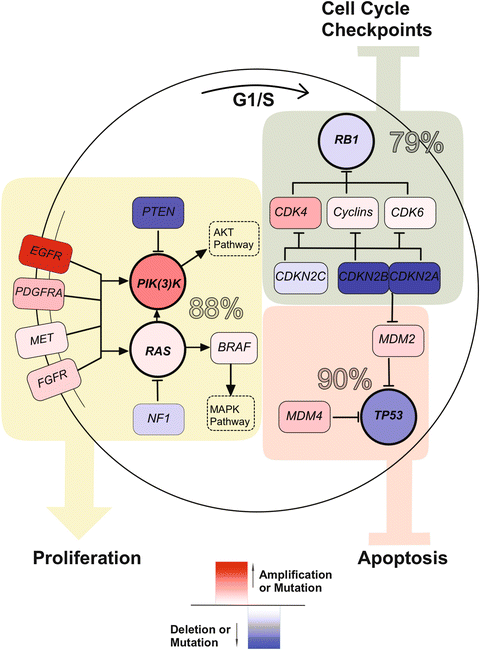
Fig. 35.3
Core defects in three signaling pathways involved in the pathogenesis of Glioblastoma . Overall alteration rate is summarized for the TP53 pathway (eluding apoptosis), PI(3)K/RAS pathway (increasing proliferation), and RB1 pathway (avoiding cell cycle checkpoints).
35.3.8 Medulloblastoma
Medulloblastomas are considered embryonal tumors arising from the dorsal brainstem or the cerebellum. Progenitor cells from the cochlear nucleus, dorsal brainstem, and neuron precursors from the external granular layer of the cerebellar germinal zone are the proposed cells of origin [165]. Medulloblastomas exhibit a high amount of heterogeneity at histomorphological and subcellular level resulting in variable clinical behavior. Histological subtypes comprise classical (70–80 %), nodular desmoplastic/extensive nodular (16 %) and large cell anaplastic (10 %), while all are classified as WHO Grade IV [9, 166]. Over the past few years novel molecular subclassifications have been established that put histopathologic differentiations into perspective.
35.3.9 Chromosomal Alterations
Almost all medulloblastoma samples show various numerical gains or losses of chromosomal regions. The most common specific cytogenetic abnormality seen in this tumor type is isochromosome 17q (iso17q), which occurs in about 50 % of medulloblastomas [167, 168]. The breakpoint is often in the proximal p-arm (17p11), producing a dicentromeric isochromosome. Loss of one arm of chromosome 17p (25–35 %) is often a consequence of isochromosome 17q formation, but may also occur in isolation [169, 170]. In certain cases, smaller deletions are seen, with the minimal deleted region occurring at 17p13.3 [171]. The frequent involvement of 17p in medulloblastoma has resulted in efforts to identify the targeted putative tumor suppressor gene (TSG). REN(KCTD11) maps to 17p13.2, and is a candidate TSG for medulloblastoma on chromosome 17p [172]. Aberrations of chromosomal regions have also been evaluated as prognostic markers, whereas iso17q and losses of 10q and 17p are correlated with a poor prognosis and loss of chromosome 6 (monosomy 6) with good outcome across all subgroups [173]. Loss of chromosome 9q, which contains the suppressor gene locus PTCH 1, promotes activation of Sonic Hedgehog (SHH) signaling pathway and is used as a co-determinant of the SHH molecular subgroup [11].
35.3.10 Gene Amplification and Overexpression
Gene amplifications are relatively infrequent in medulloblastoma (<10 %), and typically involve the MYC and MYCN proto-oncogenes [166]. Such amplifications have been observed in the context of double minute chromosomes [174]. Amplification and/or overexpression of MYC-family genes are regularly observed in the large cell anaplastic variant of medulloblastoma, and correlate with poor clinical outcome [175, 176]. In addition to gene amplifications, certain genes are aberrantly overexpressed in medulloblastoma such as ERBB2 [177]. Increased ERBB2 and ERBB4 expression correlates with higher risk of metastasis and is associated with a poor prognosis [178–180].
35.3.11 Gene Mutations and Deletions
Various mutations of tumor suppressor genes important in developmental signaling pathways have been described in medulloblastoma. Mutations in PTCH1, SMOH, Gli2, and SUFU lead to loss of inhibitory factors and thus promote constitutive activation of SHH pathways [181–184]. Germ-line APC mutations as found in familial adenomatous polyposis (FAP) syndrome or Turcot syndrome predispose to medulloblastoma formation [86]. The APC-protein is encoded on chromosome 5 and is part of a protein complex inhibiting the WNT signaling pathway. Loss of APC function results in intranuclear accumulation of ß-catenin, which activates transcription factors and promotes the pathogenesis of medulloblastoma. Overall, mutations involving the WNT pathway occur in approximately 10 % of sporadic medulloblastoma cases, including activating mutations in the CTNNB1 gene [185–188]. In addition, TP53 mutations have been found in 10 % of medulloblastoma cases (16 % in WNT, 21 % in SHH, virtually absent in Group 3 and 4) and are considered a risk factor for poor outcome in the SHH subgroup [189].
35.3.12 Epigenetic Alterations
Epigenetic alterations such as modification of histone or DNA methylation status are important factors in medulloblastoma pathogenesis and mostly recorded in Group 3 and 4 medulloblastoma [190, 191]. Large-scale genomic analyses have identified OTX2, a developmentally regulated transcription factor important for early brain morphogenesis, as a frequently occurring focal genetic gain in medulloblastoma (21 %) [168, 192, 193]. Overexpression of OTX2 results in impaired demethylation activity of histone modifying proteins. Consequently, levels of trimethylated histone H3 lysine 27 (H3K27me3) are increased which in turn leads to specific chromatin condensation [194]. Medulloblastoma patients with OTX2 gene amplification (mostly restricted to subgroup 3 and 4) exhibited poorer survival and were more likely to have anaplastic tumors [193]. There is also epigenetic deregulation of DNA methylation as an underlying mechanism of medulloblastoma formation. Subgroup specific patterns of DNA methylation have already been proposed as a biomarker for improving survival prediction in non-WNT medulloblastoma [195, 196].
35.3.13 Subgroups
Advances in next generation DNA sequencing and ultrahigh-resolution genetic mapping over recent years have enabled remarkable insight into some of the underlying molecular interactions leading to the observed heterogeneity in clinical behavior. According to current consensus among investigators, four main subgroups can be distinguished: WNT, SHH, Group 3, and Group 4 [11, 197, 198] (Fig. 35.4). The WNT and the SHH group are marked by mutations in the WNT and SHH pathway, respectively. For Group 3 and 4, no single pathway alteration has been identified and the diagnosis relies primarily on transcriptional profile clusters. However, recent studies detected prevalent drivers in particular for Group 3 [199]. These molecular subgroups typically do not correlate with the histological phenotypes. The histopathological classic type occurs in all four medulloblastoma subgroups. The nodular desmoplastic or extensive nodular type falls within the SHH subtype, although it should be mentioned that the SHH subgroup is the only subgroup that may include all of the known major histological variants. Large cell anaplastic tumors are also found in all four subgroups but the majority will be classified to Group 3. The WNT subgroup has mostly classic histology and accounts for 10 % of all medulloblastomas and has the best prognosis with >90 % long-term survivals. Alterations resulting in increased activity of the WNT pathway results from sporadic somatic mutations of CTNNB1 encoding ß-catenin or less common in germ-line mutations of the APC gene as in Turcot or FAP syndrome. Monosomy 6 and TP53 mutation are often found. The SHH subgroup (30 %) has a good to intermediate prognosis; almost half of the cases exhibit desmoplastic/nodular histology.
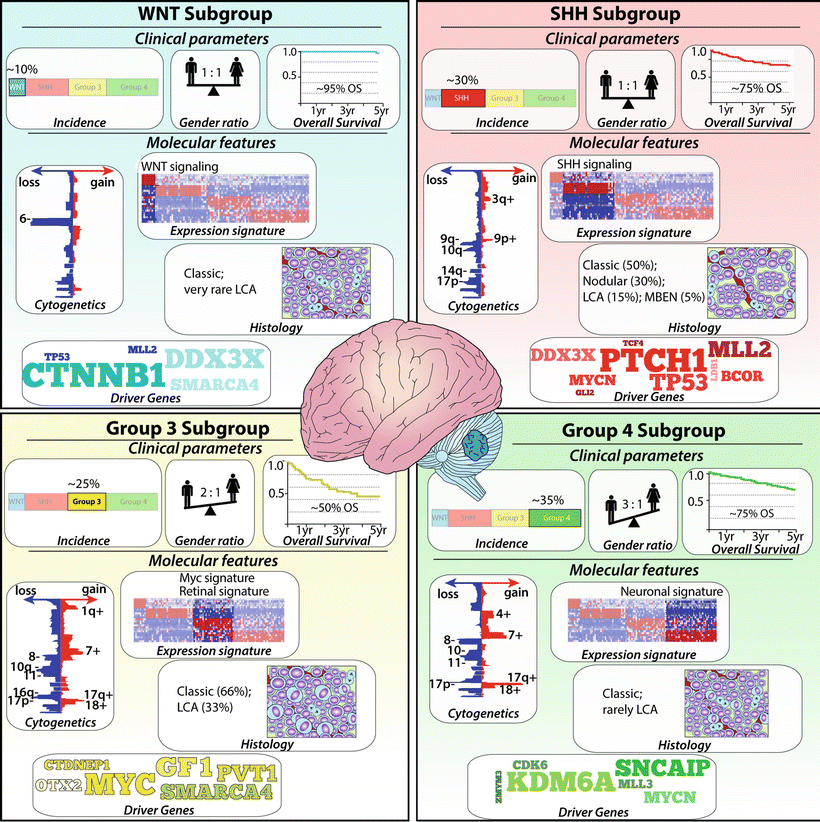
Fig. 35.4
Medulloblastoma Subgroups. Infographic illustrating the incidence, gender ratio, survival, histology, cytogenetic alterations, and common driver genes for each group. LCA large cell anaplastic, MBEN medulloblastoma with extensive nodularity.
In sporadic SHH tumors , somatic mutation in PTCH, SMO, and SUFU leads to increased SHH activity. In addition, amplifications of GLI1 and GLI2 can trigger this pathway. Deletion of chromosome 9q, which includes the PTCH gene, is primarily limited to SHH tumors. Hereditary SHH medulloblastomas are found in Gorlin syndrome and are characterized by germ-line mutation of the tumor suppressor gene PTCH1. MYCN amplification and mutations at TP53 are regularly observed. Recently, small molecules targeting smoothened (SMO), a positive regulator of the SHH pathway, showed rapid (although transient due to acquired drug resistance) regression of tumor volume in a subset of patients with SHH medulloblastoma [200, 201]. Group 3 medulloblastomas (25 %) primarily demonstrate classic histology but have a high incidence of large cell anaplastic types. They are almost never observed in adults and harbor the worst prognosis of all subtypes. Metastases are frequently found and coupled to poor prognosis. MYC and OTX2 amplification are common features and some tumors show loss of both 5q and 10q and gain of chromosome 1q. Recently, somatic genomic rearrangements resulting in GFI1 and GFI1B activation have been found to occur in approximately one-third of Group 3 patients; thus enhancer hijacking is now considered a prominent mechanism driving Group 3 medulloblastoma [199]. Group 4 is the most common medulloblastoma subgroup (35 %), usually of classic histology and with an intermediate prognosis. Isochromosome 17q is the major cytogenic alteration observed (60–80 % of Group 4 samples). Amplification of OTX2, CDK6, and MYCN are also frequently associated with this subgroup, whereas structural variants associated with GFI1 and GFI1B activation are detected in 5–10 % [199].
Molecular classification will have a large impact on future treatment recommendations and strategies. Given the four subgroups show distinct clinical behavior that does not correlate with histological features, the use of histopathology alone is considered inadequate for the classification of medulloblastoma. It remains to be seen how the revealed diversity in molecular patterns and clinical outcome will influence the WHO Grade IV classification for medulloblastoma. The current treatment modality of surgery, radiation and chemotherapy is highly toxic to the developing brain. Complication and morbidity observed in patients with tumors subgrouped into good prognosis, such as the WNT subgroup, could correspond to a higher extent with iatrogenic interventions than with the natural course of disease. These patients may benefit from a reduction in radiation and chemotherapy, whereas patient with tumors subgrouped into more aggressive lesions may benefit from exhaustive therapeutic strategies.
35.3.14 Ependymoma
Ependymomas originate from the wall of the ventricular system with possible manifestations along the entire craniospinal axis of the CNS [202]. Radial glia cells are presumed to constitute the progenitor cells [203]. In children, over 90 % of ependymomas occur intracranially with 70 % arising in the posterior fossa, whereas in adults the spinal manifestation is more common [204–206]. Based on histopathological features, the WHO divides ependymomas in grade I–III and recognizes four histological types: subependymoma (grade I), myxopapillary (grade I, usually spinal), classic (grade II), and anaplastic (grade III) [9, 202]. However, as for most tumors, molecular characteristics will have to complement and specify future grading systems. Recent studies indicate that ependymomas from different compartments of the CNS display distinct genetic signatures, reflecting their unique origins, despite appearing histologically homogeneous [203, 207].
35.3.15 Chromosomal Alterations
Cytogenetic changes are seen in a significant proportion of ependymomas and are overall more common in adults [206, 208, 209]. About 30 % of sporadic ependymomas have monosomy 22, and deletions or translocations involving chromosome 22 are also observed [210]. The NF2 gene is located on chromosome 22 (22q12), and NF2 patients are predisposed to develop spinal ependymomas in addition to other CNS tumors [211]. In fact, NF2 mutations are almost exclusively found in spinal ependymomas [212], thus another tumor suppressor gene on chromosome 22 might be involved in cranial ependymomas [210, 213]. Gain of chromosome 1q is the most common genomic aberration in pediatric intracranial ependymomas, possibly associated with anaplastic histology [206, 214]. Besides numeric chromosomal alterations within the ependymoma genome, translocations are frequently reported to affect chromosomes 1, 11, and 22 [208]. The assumption that tumor location-specific genomic alterations exist in ependymoma has been encouraged by recent revelation of chromothripsis within chromosome 11q13.1 [215]. In more than two-thirds of supratentorial ependymoma samples, a fusion of the oncogene RELA to an uncharacterized gene involving 11q13.1 occurs, which is absent in posterior fossa tumors. The aberrant RELA-fusion proteins modulate activation of the transcription factor NF-kB. The largest molecular analysis of ependymoma to date, including 583 tissue samples, demonstrated that posterior fossa ependymomas can be classified in to two subgroups, Group A and B, which differ in their genomics, transcriptomics, location and clinical outcome [209]. Group A ependymomas affect younger patients, are more laterally located and exhibit more frequent recurrence and metastasis with an overall poorer prognosis compared with Group B. The genome in Group A is more balanced, shows increased occurrence of chromosome 1q gains, whereas in Group B the genome is highly unstable with frequent segmental gains of chromosomes 9, 15, 18, as well as losses of chromosomes 6 and 22 [209].
35.3.16 Gene Amplification, Overexpression, Mutations, and Deletions
In ependymoma and especially in children, the overall genetic mutation rate appears to be lower than astrocytic tumors. Expression profiling by microarray analysis on pediatric ependymomas have identified a subset of genes abnormally expressed in this tumor, including increased expression of EGFR, WNT5A, TP53, and many cell cycle, cell adhesion, angiogenesis, and cell proliferation genes, with downregulated genes including SCHIP-1 and EB1 [213]. EGFR overexpression has been recorded as a prognostic marker in intracranial grade II ependymomas correlating with reduced progression free and overall survival [216, 217]. Amplification and rearrangement of the c-erb B gene has been identified, as well as rearrangements of the MEN1 gene (11q13) [212, 218]. A large genomic analysis of ependymoma including tumors from 207 samples demonstrated focal amplification of genes involved in stem cell proliferation, pluripotency, and neuronal differentiation such as THAP11, PSPH, EPHB2, KCNN1, RAB3A, PTPRN2, PCDH cluster, and NOTCH1 [207]. This same study identified focal deletions of known tumor suppressor genes (PTEN, INK4A/ARF) and novel genes such as STAG1 and TNRC6B [207].
35.3.17 Epigenetic Alteration
Epigenetic modifiers involved in the formation of ependymomas have recently been identified. The methylation status of CpG islands occurs more frequently in posterior fossa Group A (PFA) tumors, leading to transcriptional silencing of the histone methyltransferase Polycomb repressive complex 2 (PRC2), which in turn results in repressive effect on expression of cell differentiation genes [219]. This epigenetic mechanism predicts for the more aggressive behavior of Group A tumors. Mice xenograft models implanted with human PFA showed a decreased tumor volume and longer survival after that PRC2 was reactivated through demethylation by administration of established epigenetic drugs that target either DNA or histone methylation [219].
35.4 Cell Cycle Dysregulation and Mitogenic Factors in Human Brain Tumors
The cell cycle can be conceptualized as a circle divided by a number of discrete phases during which nuclear content changes in preparation for cell division (Fig. 35.5). In healthy neural tissue, with advancing differentiation and age, most cells will become quiescent and do not express cell cycle genes due to the presence of cell-type specific repression mechanisms. In human brain tumors, the intrinsic machinery of the cell cycle is the target of mutations or functional derangements leading to release of repression mechanisms and rapid, uncontrolled proliferation—a hallmark of cancer.