Genes in bold are present in more than one histological subtype.
Data generated through of analysis of 183 lung adenocarcinomas24; TCGA project of 178 previously untreated, stage I-IV primary lung squamous cell carcinoma9; and 34 primary SCLC tumors and 17 SCLC cell lines.25
Identification of Novel Pathways
TCGA project found a significant number of lung SCCs had alterations in genes involved in oxidative stress response and squamous differentiation. Genomic alterations included point mutations and copy number alterations. More specifically, alterations in one of the three genes NFE2L2, KEAP1, and CUL3 were identified in nearly a third of tumor samples studied. The master antioxidant transcription factor NFE2L2 promotes survival following cellular insults that trigger oxidative damage and is regulated by KEAP1, an oxidative stress sensor. In unstressed conditions, KEAP1 binds and subsequently represses NFE2L2. KEAP1 also forms a ubiquitin E3 ligase complex with CUL3, resulting in constant ubiquitination of NFE2L2. Mutations in NF2L2 occurred nearly exclusively in one of the two KEAP1 interaction motifs. Mutations in KEAP1 and CUL3 showed a pattern consistent with loss of function. In addition, mutations in KEAP1 and CUL2 were mutually exclusive with NFE2L2. Alterations in genes that are known to play a role in squamous differentiation were identified in 44% of lung SCC samples. The changes include overexpression and amplification of SOX2 and TP63 and loss-of-function mutations involving NOTCH1 and NOTCH2. Truncating mutations involving NOTCH1 and NOTCH2 have been reported previously in squamous cell cancer of the skin and lung.
Recurrent somatic mutations in the splicing factor gene U2AF1, truncating mutations affecting RBM10 and ARID1A, and in-frame exonic alterations within EGFR and SIK2 kinases were identified in an exome and genome analysis of lung adenocarcinoma. SOX2 mutations and amplification and a recurrent RFL-MYCL1 fusion were common in an exome, transcriptome, and copy number analysis of 34 primary SCLC tumors and 17 SCLC cell lines. Suppression of SOX2 in SOX2-amplified cell lines or MYCL1 in RLF-MYCL1 cell lines both resulted in decreased proliferation, suggesting that these alterations may represent SCLC subtype vulnerabilities.
Identification of Therapeutic Targets
The lung SCC TCGA project reported a number of potentially targetable alterations using a gene-centered and pathway-directed approach. Using fairly stringent criteria (availability of a targeted agent, confirmation of altered allele in transcriptome sequencing, and Mutation Assessor Score), a potentially targetable gene was identified in 64% of samples studied. Alterations in one of the three core pathways (PI3K/AKT/mTOR, RTKs, and RAS/RAF/MAPK) were found in 69% of samples even after restricting the analysis to include only those where mutations were confirmed by transcriptome sequencing and those amplifications associated with overexpression of the target gene. Some of the notable targets altered include PI3KCA, PTEN, AKT3, BRAF, FGFR, and EGFR. Another novel target identified with whole-transcriptome analyses of tumor samples is in-frame fusion transcripts involving KIF5B (the Kinesin family 5B gene) and the RET oncogene, which is found in 1% to 2% of patients with lung adenocarcinoma and is discussed later.
Lessons Learned and Future Directions
Preliminary TCGA analysis of lung SCCs has demonstrated the importance of integrating mutational data with other genomic data such as methylation, mRNA expression, and copy number. CDKN2A, a tumor suppressor gene (TSG) that encodes two cell cycle inhibitor proteins, p16 and p14, is frequently altered in lung SCC. CDKN2A is inactivated through multiple mechanisms from epigenetic silencing by methylation (21%), to inactivating mutation (18%), to other events such as exon skipping (4%) and homozygous deletion (29%). Thus considering only one set of genomic data could lead to inaccurate conclusions on the role of the gene.
It is clear that next-generation sequencing has enormous potential to unravel the complexities of the lung cancer genome and identify the molecular mechanisms underpinning therapeutic responses and progression of lung cancer. Although the challenges in gathering reliable and clinically and pathologically annotated data are not trivial, high-throughput technologies and publicly stored genome-wide databases related to lung cancer are resources with the potential to drive a global collaborative effort in identifying new targets for lung cancer diagnostics and therapeutics. Large-scale multidisciplinary and international collaborations such as the TCGA project, the NCI Lung Cancer Mutation Consortium (LCMC),10as well as international lung cancer sequencing consortiums will enable the uniting of clinically annotated with molecularly annotated lung cancer specimens. Enabling free access to all of these genome-wide studies will allow independent confirmation on the role of the various molecular changes for prognosis, prediction, and targeting of therapy of lung cancer.
“Synthetic lethal” screens using RNAi (siRNAs and shRNA libraries) technology have allowed unbiased, genome-wide approaches to identification of genes whose perturbation can selectively kill lung cancer cells. The ability to identify “synthetic lethality”11associated with oncogenic changes in tumor cells has particular utility in identifying new therapeutic targets or molecules to treat traditionally hard-to-target tumors, such as those with oncogenic KRAS. Small interfering RNA (siRNA) and short-hairpin RNA (shRNA) screens have identified genes whose perturbation can selectively sensitize NSCLC cell lines to sublethal doses of chemotherapeutic agents, sensitize KRAS mutant cells to targeted drugs, suppress tumorigenicity in cells with specific gene dysregulation such as oncogenic KRAS or aberrant EGFR, and identify novel genes critical for tumorigenic processes such as metastasis.
Epigenetic Changes in Lung Carcinogenesis
Epigenetic events can lead to changes in gene expression without any changes in DNA sequence and therefore, importantly, are potentially reversible.
Methylation and Histone Modification
Aberrant promoter hypermethylation (the addition of a methyl group to CpG islands in the promoter region of a gene that results in transcriptional silencing) is a common method for inactivation of TSGs in tumor cells and occurs early in lung tumorigenesis. In fact, whole-genome microarray profiling of DNA methylation patterns in lung cancer—termed the lung cancer epigenome or methylome—suggests that the role of methylation in lung tumorigenesis may have been previously underestimated. Because aberrant methylation is an early event in lung cancer pathogenesis and is detectable in DNA circulating in the blood, many studies have investigated methylation status as a biomarker for risk assessment, early detection, disease progression, and prognosis in lung cancer (Table 32-3 ).
DNA is methylated by DNA methyltransferases (DNMTs) which are responsible for both de novo and maintenance of preexisting methylation in a cell. Histone modification is another mechanism for epigenetic control of gene transcription: histone deacetylation results in condensing of chromatin, resulting in transcriptionally inactive DNA. Inhibitors of DNMTs or histone deacetylases (HDACs) resulting in pharmacologic restoration of expression of epigenetically silenced genes is an exciting targeted therapeutic approach and shows promise in lung cancer (Table 32-4 ).
microRNA-Mediated Regulation
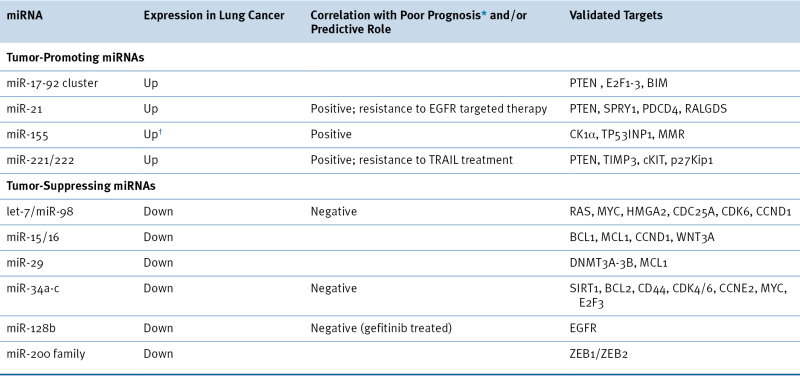
There is currently a strong research focus on microRNAs (miRNAs) as potential diagnostic and prognostic biomarkers and therapeutic targets for lung cancer. miRNA profiles for histologic and prognostic classification of lung tumors and detection of miRNAs in peripheral blood and sputum illustrate the potential of miRNAs as diagnostic and early detection biomarkers in lung cancer. Table 32-5 summarizes some experimentally validated miRNAs important in lung cancer. miRNAs are a class of non–protein-encoding small RNAs capable of regulating gene expression by either directly cleaving a targeted mRNA or inhibiting translation by interacting with the 3′ untranslated region (UTR) of a target mRNA. A single miRNA often targets multiple genes, and multiple miRNAs may target the same mRNA, which results in a complex network of molecular pathways where a single miRNA (to date, more than 1400 human miRNAs have been identified) can potentially affect multiple cellular processes. Aberrant expression of miRNAs has been found to play an important role in the pathogenesis of lung cancer as either oncogenes or TSGs. miRNAs can function as either TSGs or oncogenes. Restoration of aberrantly expressed miRNAs can be achieved in vitro and in vivo using miRNA mimics (for underexpressed miRNAs) or miRNA inhibitors (termed antisense oligonucleotides or antagomirs; for overexpressed miRNAs). Concurrent inhibition or overexpression of miRNAs with conventional therapies has resulted in an increased response to EGFR TKIs, radiotherapy, and chemotherapy. These studies illustrate the potential of miRNAs in lung cancer therapeutics development; however, limitations in pharmacokinetics, delivery, and toxicity need to be addressed.
Data summarized from the following reviews: References 26–28.
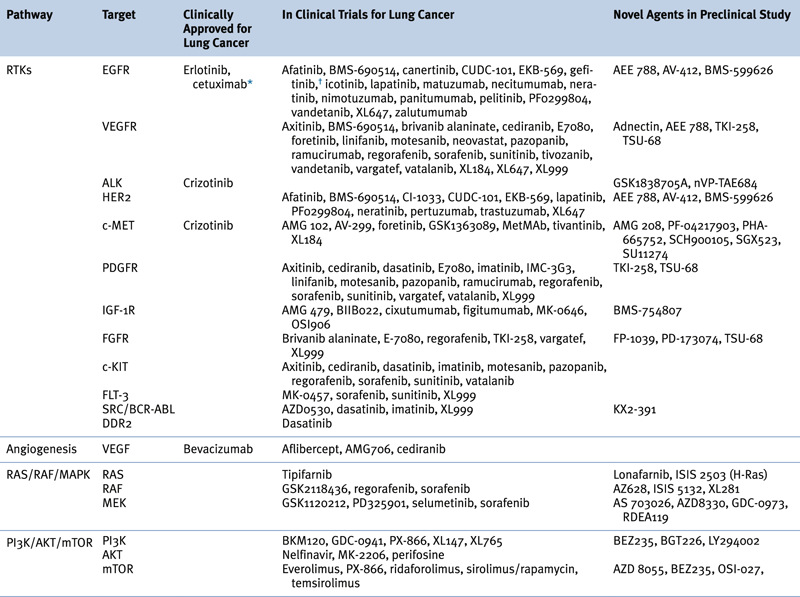
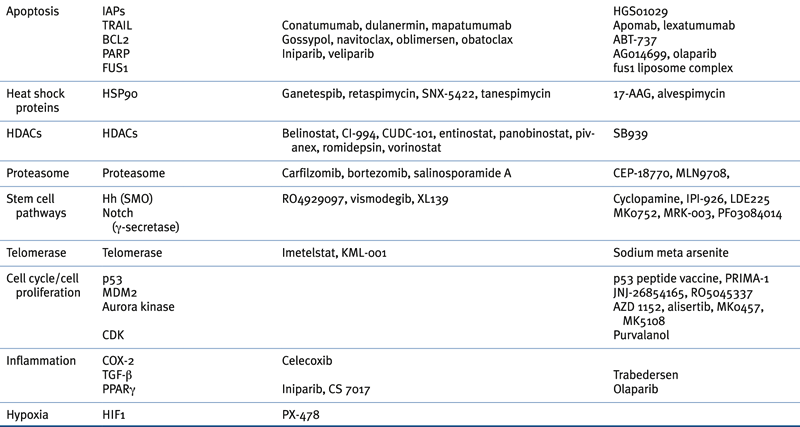
Table 32-4
Targeted Therapies Approved in Clinical Trial or in Preclinical Study for Lung Cancer
∗Although not currently U.S. FDA-approved for non–small-cell lung cancer, cetuximab is a recommended treatment in several practice guidelines, including those of the American Society of Clinical Oncology (ASCO) and the National Comprehensive Cancer Network (NCCN).
†Previously approved in the United States and still approved elsewhere.
Data summarized from the following reviews: References 31–34.
The let-7 family is a cluster of miRNAs that function as tumor suppressors and is frequently underexpressed in lung tumors, particularly NSCLC, compared to normal lung, and decreased expression has been associated with poorer prognosis. Let-7 regulates multiple oncogenes including RAS, MYC, and HMGA2 via binding to the let-7 binding sites in their respective 3′ UTRs. Let-7 replacement therapy shows potential, with reduced tumor burden observed in vivo; however, tumor response in patients will be affected by a SNP in the let-7 complementary site (LCS6) of KRAS, which is significantly associated with lung cancer risk and results in increased KRAS expression in vitro.
An example of an important oncogenic miRNA—oncomir—in lung cancer is RAS-regulated miR-21 which promotes cellular growth and invasion and metastasis by targeting multiple genes with tumor suppressive effects such as negative regulators of the RAS/RAF/MAPK pathway, proapoptotic, and anti-metastatic genes. Expression of miR-21 is also suggested to be positively regulated by the EGFR signaling pathway, specifically EGFR mutations. Some miRNAs have also been shown to be important mediators of metastasis. The expression of miR-200 family members is commonly lost in aggressive lung cancers and can prevent epithelial to mesenchymal transition (EMT)—and consequently, invasion and metastasis—by repressing transcriptional repressors of E-cadherin.
Oncogenes, Tumor Suppressor Genes, and Signaling Pathways in Lung Cancer
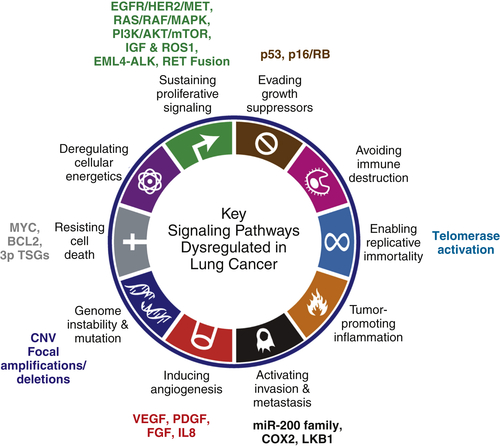
The “hallmarks of cancer”4describe the complexities of neoplastic disease and stratify the complexities by mechanistic function. Genomic instability is an underlying “enabling” characteristic of lung cancer cells where alterations such as chromosomal rearrangements can generate rare genetic events in cells that eventually give rise to cancer. Mapping amplifications and deletions in copy number throughout the cancer genome has led to the identification of many oncogenes and TSGs. Recent whole-genome genomic approaches have yielded further insight into the complexities of the lung cancer genome with the identification of driving mutations and other key signaling pathways (see Table 32-2). The following section summarizes known driver mutations (EGFR, KRAS, and EML4-ALK) and key signaling pathways (including RAS/RAF/MAPK, PI3K/AKT/mTOR, p53, and p16/RB) in lung cancer organized by “hallmarks” (Figure 32-1 ).12There are several targeted therapy agents in the clinic or in development for lung cancer (see Table 32-4).
Figure 32-1Key signaling pathways discussed in this chapter that are commonly dysregulated in lung cancer in relation to the “Hallmarks of Cancer” proposed by Hanahan and Weinberg4Currently, most of our knowledge of the molecular changes in lung cancer converges on the six original hallmarks (sustaining proliferative signaling; evading growth suppressors; resisting cell death; enabling replicative immortality; inducing angiogenesis; and activating invasion and metastasis), as well as the newly categorized “enabling characteristic” genome instability and mutation.(Reprinted from Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674, with permission from Elsevier.)
Oncogene activation probably occurs in all lung cancers (typically by gene amplification, overexpression, point mutation, or DNA rearrangements), resulting in persistent upregulation of mitogenic growth signals, which induce cell growth. Importantly, it can also result in “oncogene addiction” in which the cell becomes dependent on this aberrant oncogenic signaling for survival.13These “driver” oncogenes or oncogene “addictions” represent acquired vulnerabilities in lung cancer cells and present as significant therapeutic targets by offering the specificity of killing tumor but not normal cells.
Loss of TSG function is an important step in lung carcinogenesis and usually results from inactivation of both alleles, with loss of heterozygosity (LOH; through chromosomal deletion or translocation) inactivating one allele, and point mutation or epigenetic or transcriptional silencing inactivating the second allele. Commonly inactivated TSGs in lung cancer include TP53, RB1, STK11, CDKN2A, FHIT, RASSF1A, and PTEN. Historically, tumor suppressors have been more difficult to target with therapeutic agents because restoration of lost activity is much more difficult than inhibition of increased activity (as with oncogenes), and consequently most endeavors were targeted at downstream effectors. Increased understanding of the function of tumor suppressor proteins may identify novel therapeutic targets, as shown with p53, where compounds that stabilize the mutant protein or restore wild-type conformation demonstrate clinical utility.
Hallmark: Sustaining Proliferative Signaling
EGFR/HER2/MET Signaling
EGFR
The ErbB family of tyrosine kinase receptors includes four members: EGFR, ERBB2 (HER2), ERBB3, and ERBB4. The receptors can activate multiple signal transduction cascades, including the RAS/RAF/MAPK, PI3K/AKT/mTOR, and STAT pathways, by forming homo- and heterodimers with different ligand specificity. EGFR is overexpressed or aberrantly activated in 50% to 90% of NSCLCs. EGFR-targeted inhibitors include monoclonal antibodies that target the EGFR extracellular domain and tyrosine kinase inhibitors (TKIs), which are small molecules that inhibit intracellular tyrosine kinase activity of EGFR. A significant advance was made in the treatment of NSCLC with the observation that lung cancer somatic mutations in the kinase domain of EGFR strongly correlated with sensitivity to EGFR TKIs. EGFR mutant lung tumors exhibit exquisite sensitivity and marked tumor response with EGFR TKIs (such as erlotinib and gefitinib) and antibodies (such as cetuximab)—an example of oncogene addiction in lung cancer where tumors driven by EGFR mutation-activation of EGF signaling rely on continued EGF signaling for survival. “Classic” EGFR mutations in the tyrosine kinase domain (by either exon 19 deletion or exon 21 L858R mutation, which each account for about 45% of EGFR mutations) show an increased amount and duration of EGFR activation compared with wild-type receptors and have preferential activation of the PI3K/AKT/mTOR and STAT3/STAT5 pathways rather than the RAS/RAF/MAPK pathway. By contrast, the remaining 10% of EGFR tyrosine kinase mutations, in exons 18 and 20, do not confer sensitivity to EGFR TKIs and in some cases are associated with EGFR TKI resistance. EGFR mutations of all types are particularly prevalent in certain patient subgroups: adenocarcinoma histology, women, never smokers, and East Asian ethnicity. Despite an initial response, patients treated with EGFR TKIs eventually develop resistance to TKIs that is linked (in approximately 50% of tumors) to a T790M mutation in EGFR exon 20. In such cases, it is likely that a small population of cancer cells harboring T790M mutations is present at diagnosis and selected for during EGFR TKI therapy. Proposed mechanisms of the T790M-associated therapeutic resistance include a conformational change resulting in steric hindrance to EGFR TKI binding and increased EGFR affinity for ATP. Resistance to TKI therapy has also been associated with EGFR exon 20 insertions; KRAS mutation; amplification or activation of the MET proto-oncogene, which provides an alternative signaling pathway; and occasionally a switch of tumor differentiation to an SCLC-like phenotype. Second-generation EGFR TKIs (such as PF00299804, afatinib, and neratinib) bind irreversibly to EGFR tyrosine kinase, induce much less therapeutic resistance, and appear effective against secondary resistance mutations such as T790M.
ERBB2 (HER2)
The ligand for HER2 remains unknown, but HER2 is activated following homo- or heterodimerization (with EGFR or HER3 preferentially). Unlike breast and gastric cancers, HER2 amplification or overexpression in NSCLC does not confer sensitivity to HER2 antibodies or TKIs. However, exon 20 mutations in HER2 mutations (occurring in 3% to 10% of lung adenocarcinomas) do confer sensitivity to lapatinib in NSCLC cell lines. HER2 mutations also confer resistance to EGFR TKIs regardless of EGFR mutation status as HER2 replaces EGFR in driving growth signals.
MET
Similar to EGFR, MET is a receptor tyrosine kinase capable of driving RAS/RAF/MAPK and PI3K/AKT/mTOR pathway signaling following activation on hepatocyte growth factor (HGF) binding. Amplification of MET is also thought to mediate resistance to EGFR TKIs, independent of the T790M mutation, where MET activates the PI3K/AKT/mTOR pathway through phosphorylation of HER3, independent of EGFR and HER2. Inhibition of MET is being successfully approached with antibodies (such as MetMAb) and small-molecule MET inhibitors (tivantinib/ARQ-197) (see Table 32-4).
RAS/RAF/MAPK Pathway
The RAS proto-oncogene family (KRAS, HRAS, NRAS, and RRAS) encodes four highly homologous 21-kDa membrane-bound proteins involved in signal transduction. Activation of the RAS/RAF/MAPK pathway occurs frequently in lung cancer, most commonly via activating mutations in KRAS (approximately 20%, particularly adenocarcinomas). In lung cancer, 90% of mutations are located in KRAS (80% in codon 12, and the remainder in codons 13 and 61), with HRAS and NRAS mutations only occasionally documented. Proteins encoded by the RAS genes exist in two states: an active state, in which GTP is bound to the molecule, and an inactive state, where the GTP has been cleaved to GDP. Activating point mutations confer oncogenic potential through loss of intrinsic GTPase activity, resulting in an inability to cleave GTP to GDP. This results in constitutive activation of downstream signaling pathways, such as PI3K and MAPK, rendering KRAS mutant tumors independent of EGFR signaling and therefore resistant to EGFR TKIs as well as chemotherapy. KRAS mutations are mutually exclusive with EGFR and ERBB2 mutations and are primarily observed in lung adenocarcinomas of smokers. The prevalence and importance of KRAS in lung tumorigenesis make it an attractive therapeutic target. Two unsuccessful approaches were farnesyltransferase inhibitors, to inhibit posttranslational processing and membrane localization of RAS proteins, and antisense oligonucleotides against RAS. More recently, efforts have been centered on downstream effectors of RAS signaling: RAF kinase and mitogen-activated protein kinase (MAPK) kinase (MEK).
BRAF is the direct effector of RAS. Although it is commonly mutated in melanoma (about 70%), mutations are rare in lung cancer (about 3%), predominantly in adenocarcinoma, and mutually exclusive to EGFR and KRAS mutations. Strategies to inhibit RAF kinase include degradation of RAF1 mRNA through antisense oligodeoxyribonucleotides, and inhibition of kinase activity with small molecule kinase inhibitors such as the multikinase inhibitor sorafenib (which inhibits VEGFR, PDGFR, FLT-3, RAF, MEK, and KIT) as well as some BRAF mutant-specific inhibitors such as vemurafenib, PLX-4720, and GDC-0879. Several potent and selective MEK inhibitors such as selumetinib (AZD6244) and GSK1120212 show potential in inhibiting RAS/RAF/MAPK signaling (see Table 32-4). Attempts to directly inhibit or perturb mutant KRAS continue with the advent of whole-genome approaches. Synthetic lethal siRNA screens have identified siRNAs that specifically kill human lung cancer cells with KRAS mutations in vitro. In addition, the combination of anti-KRAS strategies (such as depletion with shRNAs) with other targeted drugs has shown potential therapeutic utility.
PI3K/AKT/mTOR Pathway
Phosphoinositide 3-kinases (PI3Ks) are lipid kinases that regulate cellular processes such as proliferation, survival, adhesion, and motility. The PI3K/AKT/mammalian target of rapamycin (mTOR) pathway is downstream of several receptor tyrosine kinases including EGFR, and downstream effectors are involved in cell growth, angiogenesis, cell metabolism, protein synthesis, and suppression of apoptosis directly or via the activation of mTOR. In lung tumorigenesis, activation of the PI3K/AKT/mTOR pathway occurs early in pathogenesis, generally through mutations or amplification of (oncogenes) PI3K (as well as EGFR or KRAS), activation of AKT, or PTEN loss of function (TSG), and promotes cell survival through inhibition of apoptosis. PTEN, TSC1, TSC2, and STK11 (LKB1) are tumor suppressors that function as negative regulators of the pathway, and thus their loss of function activates the pathway. PTEN antagonizes the PI3K/AKT/mTOR pathway by dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3), a product of PI3K, to PIP2 and is commonly inactivated in lung cancer by mutations or loss of expression. TSC1 and TSC2 form a complex that inhibits activity of small G protein Rheb, leading to inhibition of the mTOR complex mTORC1. TSC1/TSC2-mediated inhibition of mTORC1 can be activated by LKB1 and AMPK and inhibited by AKT-mediated phosphorylation of TSC2. The serine/threonine kinase mTOR, a downstream effector of AKT, is an important intracellular signaling enzyme in the regulation of cell growth, motility, and survival in tumor cells. Molecular characterization of PI3K/AKT/mTOR pathway biomarkers (such as loss of PTEN) will enable better selection of tumors responsive to mTOR, AKT, and PI3K inhibition.
Only gold members can continue reading. Log In or Register to continue