Melanoma Therapy and Molecular Targets
Mapk-Braf and Intersection of Other Pathways
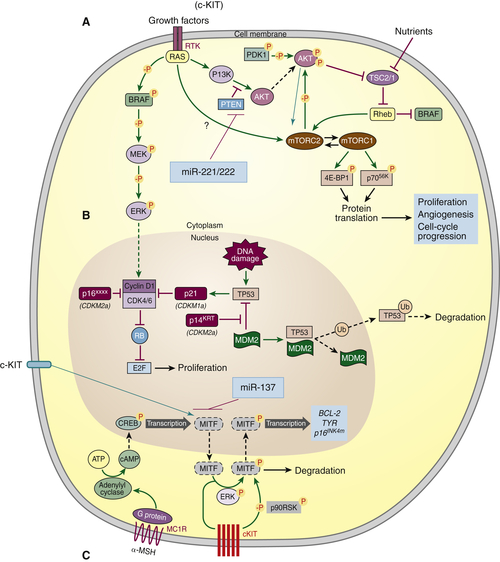
c-Kit
Epigenetic Pathways: MicroRNA
Paratumoral Pathways
Conclusions
1. DNA contents of chromosome Ph1 and chromosome 21 in human chronic granulocytic leukemia . Science . 1964 ; 144 : 1229 – 1231 .
2. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining . Nature . 1973 ; 243 : 290 – 293 .
3. A quantitative assay for transformation of bone marrow cells by Abelson murine leukemia virus . J Exp Med . 1976 ; 143 : 1453 – 1463 .
4. Identification of an Abelson murine leukemia virus-encoded protein present in transformed fibroblast and lymphoid cells . Proc Natl Acad Sci U S A . 1978 ; 75 : 2488 – 2492 .
5. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia . Nature . 1982 ; 300 : 765 – 767 .
6. The CML-specific P210 bcr/abl protein, unlike v-abl, does not transform NIH/3T3 fibroblasts . Science . 1987 ; 237 : 532 – 535 .
7. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells . Nat Med . 1996 ; 2 : 561 – 566 .
8. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992-2006 . J Am Acad Dermatol . 2011 ; 65 S17-S25, e1-3 .
9. Risk-adjusted melanoma skin cancer incidence rates in Whites (United States) . Melanoma Res . 2011 ; 21 : 535 – 540 .
10. Predicting survival outcome of localized melanoma: an electronic prediction tool based on the AJCC Melanoma Database . Ann Surg Oncol . 2010 ; 17 : 2006 – 2014 .
11. Final version of 2009 AJCC melanoma staging and classification . J Clin Oncol . 2009 ; 27 : 6199 – 6206 .
12. Mechanism of skin tanning in different racial/ethnic groups in response to ultraviolet radiation . J Invest Dermatol . 2005 ; 124 : 1326 – 1332 .
13. Type VII collagen antibody LH 7.2 identifies basement membrane characteristics of thin malignant melanomas . J Pathol . 1989 ; 157 : 243 – 247 .
14. An analysis of interobserver recognition of the histopathologic features of dysplastic nevi from a mixed group of nevomelanocytic lesions . J Am Acad Dermatol . 1992 ; 27 : 741 – 749 .
15. Dysplastic nevi in relation to superficial spreading melanoma . Cancer Epidemiol Biomarkers Prev . 1993 ; 2 : 99 – 101 .
16. Dysplastic nevus in histologic contiguity with acquired nonfamilial melanoma. Clinicopathologic experience in a 100-bed hospital . Arch Dermatol . 1987 ; 123 : 80 – 84 .
17. Distinct sets of genetic alterations in melanoma . N Engl J Med . 2005 ; 353 : 2135 – 2147 .
18. ABCDE—an evolving concept in the early detection of melanoma . Arch Dermatol . 2005 ; 141 : 1032 – 1034 .
19. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma . J Clin Oncol . 1999 ; 17 : 2745 – 2751 .
20. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684 . J Clin Oncol . 1996 ; 14 : 7 – 17 .
21. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993 . J Clin Oncol . 1999 ; 17 : 2105 – 2116 .
22. Improved survival with vemurafenib in melanoma with BRAF V600E mutation . N Engl J Med . 2011 ; 364 : 2507 – 2516 .
23. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification . J Clin Oncol . 2011 ; 29 : 2904 – 2909 .
24. Improved survival with ipilimumab in patients with metastatic melanoma . N Engl J Med . 2010 ; 363 : 711 – 723 .
25. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma . J Invest Dermatol . 2001 ; 117 : 1483 – 1489 .
26. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies . Clin Cancer Res . 2006 ; 12 : 4499 – 4505 .
27. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation . EMBO J . 1999 ; 18 : 2137 – 2148 .
28. Mutations of the BRAF gene in human cancer . Nature . 2002 ; 417 : 949 – 954 .
29. High frequency of BRAF mutations in nevi . Nat Genet . 2003 ; 33 : 19 – 20 .
30. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis . Cancer Res . 2004 ; 64 : 7099 – 7109 .
31. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis . Br J Cancer . 2006 ; 95 : 581 – 586 .
32. Phase II trial of temozolomide and sorafenib in advanced melanoma patients with or without brain metastases . Clin Cancer Res . 2009 ; 15 : 7711 – 7718 .
33. Sorafenib and dacarbazine as first-line therapy for advanced melanoma: phase I and open-label phase II studies . Br J Cancer . 2011 ; 105 : 353 – 359 .
34. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity . Proc Natl Acad Sci U S A . 2008 ; 105 : 3041 – 3046 .
35. Rational design of inhibitors that bind to inactive kinase conformations . Nat Chem Biol . 2006 ; 2 : 358 – 364 .
36. Inhibition of mutated, activated BRAF in metastatic melanoma . N Engl J Med . 2010 ; 363 : 809 – 819 .
37. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors . N Engl J Med . 2012 ; 366 : 207 – 215 .
38. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib . N Engl J Med . 2012 ; 366 : 707 – 714 .
39. BRAF(V600) Inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency . Clin Cancer Res . 2012 ; 18 : 2326 – 2335 .
40. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) . Nature . 2011 ; 480 : 387 – 390 .
41. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma . Cancer Res . 2008 ; 68 : 4853 – 4861 .
42. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation . Nature . 2010 ; 468 : 968 – 972 .
43. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation . Nature . 2010 ; 468 : 973 – 977 .
44. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling . J Clin Oncol . 2011 ; 29 : 3085 – 3096 .
45. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition . J Invest Dermatol . 2012 ; 132 : 1850 – 1859 .
46. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer . Clin Cancer Res . 2012 ; 18 : 2316 – 2325 .
47. Melanoma: molecular pathogenesis and emerging target therapies (review) . Int J Oncol . 2009 ; 34 : 1481 – 1489 .
48. Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options . Cancer Res . 2011 ; 71 : 7137 – 7140 .
49. Metformin accelerates the growth of BRAFV600E-driven melanoma by upregulating VEGF-A . Cancer Discov . 2012 ; 2 : 344 – 355 .
50. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas . Cancer Res . 2004 ; 64 : 5270 – 5282 .
51. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression . Cancer Res . 2006 ; 66 : 4182 – 4190 .
52. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1 . FASEB J . 2006 ; 20 : 1009 – 1011 .
53. Homozygous deletions within human chromosome band 9p21 in melanoma . Proc Natl Acad Sci U S A . 1992 ; 89 : 10557 – 10561 .
54. Alteration of chromosome 9p21 and/or p16 in benign and dysplastic nevi suggests a role in early melanoma progression (United States) . Cancer Causes Control . 2002 ; 13 : 675 – 682 .
55. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma . Nat Genet . 1996 ; 12 : 97 – 99 .
56. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation . J Cell Biol . 2005 ; 170 : 703 – 708 .
57. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF . PLoS One . 2008 ; 3 : e2734 .
58. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms . Clin Cancer Res . 2012 ; 18 : 2502 – 2514 .
59. Sunitinib therapy for melanoma patients with KIT mutations . Clin Cancer Res . 2012 ; 18 : 1457 – 1463 .
60. The nuclear RNase III Drosha initiates microRNA processing . Nature . 2003 ; 425 : 415 – 419 .
61. Role for a bidentate ribonuclease in the initiation step of RNA interference . Nature . 2001 ; 409 : 363 – 366 .
62. Argonaute2, a link between genetic and biochemical analyses of RNAi . Science . 2001 ; 293 : 1146 – 1150 .
63. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines . Cancer Res . 2007 ; 67 : 2456 – 2468 .
64. MicroRNAs exhibit high frequency genomic alterations in human cancer . Proc Natl Acad Sci U S A . 2006 ; 103 : 9136 – 9141 .
65. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor . Proc Natl Acad Sci U S A . 2009 ; 106 : 1814 – 1819 .
66. MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines . Cancer Res . 2008 ; 68 : 1362 – 1368 .
67. MicroRNA-221 and -222 pathway controls melanoma progression . Expert Rev Anticancer Ther . 2008 ; 8 : 1759 – 1765 .
68. MicroRNA identification in plasma and serum: a new tool to diagnose and monitor diseases . Expert Opin Biol Ther . 2009 ; 9 : 703 – 711 .
69. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples . RNA . 2007 ; 13 : 1668 – 1674 .
70. MicroRNA-15b represents an independent prognostic parameter and is correlated with tumor cell proliferation and apoptosis in malignant melanoma . Int J Cancer . 2010 ; 126 : 2553 – 2562 .
71. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression . Oncol Rep . 2009 ; 22 : 563 – 567 .
72. MicroRNA-193b represses cell proliferation and regulates cyclin D1 in melanoma . Am J Pathol . 2010 ; 176 : 2520 – 2529 .
73. Fibroblasts contribute to melanoma tumor growth and drug resistance . Mol Pharm . 2011 ; 8 : 2039 – 2049 .
74. Interferon alfa-2a therapy for life-threatening hemangiomas of infancy . N Engl J Med . 1992 ; 326 : 1456 – 1463 .
75. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma . J Clin Oncol . 2012 ; 30 : 34 – 41 .
76. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438) . Clin Cancer Res . 2012 ; 18 : 1129 – 1137 .
77. Adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma: a phase III randomized controlled trial of health-related quality of life and symptoms by the European Organisation for Research and Treatment of Cancer Melanoma Group . J Clin Oncol . 2009 ; 27 : 2916 – 2923 .
78. Cytokine working group study of lymphodepleting chemotherapy, interleukin-2, and granulocyte-macrophage colony-stimulating factor in patients with metastatic melanoma: clinical outcomes and peripheral-blood cell recovery . J Clin Oncol . 2010 ; 28 : 1196 – 1202 .
79. A new member of the immunoglobulin superfamily—CTLA-4 . Nature . 1987 ; 328 : 267 – 270 .
80. The yin and yang of T cell costimulation . Science . 1995 ; 270 : 932 – 933 .
81. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma . Proc Natl Acad Sci U S A . 2003 ; 100 : 8372 – 8377 .
82. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206 . J Clin Oncol . 2005 ; 23 : 8968 – 8977 .
83. Self-recognition and tumor response to immunotherapy . J Clin Oncol . 2005 ; 23 : 5875 – 5877 .
84. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma . N Engl J Med . 2011 ; 364 : 2517 – 2526 .
85. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial . Lancet Oncol . 2012 ; 13 : 459 – 465 .
86. T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells . J Immunol . 2011 ; 187 : 5233 – 5245 .
87. Molecular pathway profiling of T lymphocyte signal transduction pathways; Th1 and Th2 genomic fingerprints are defined by TCR and CD28-mediated signaling . BMC Immunol . 2012 ; 13 : 12 .
88. T-cell regulation by CD28 and CTLA-4 . Nat Rev Immunol . 2001 ; 1 : 220 – 228 .
89. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling . Nat Rev Immunol . 2003 ; 3 : 544 – 556 .
90. Treatment sequencing, asymmetry, and uncertainty: protocol strategies for combination chemotherapy . Cancer Res . 1986 ; 46 : 3876 – 3885 .
91. Rationale for the use of alternating non-cross-resistant chemotherapy . Cancer Treat Rep . 1982 ; 66 : 439 – 449 .
92. Alternating drug combinations in the treatment of advanced Hodgkin’s disease . N Engl J Med . 1982 ; 306 : 770 – 775 .
93. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial . Lancet . 2007 ; 369 : 29 – 36 .
94. Clinical and immunologic effects of intranodal autologous tumor lysate-dendritic cell vaccine with aldesleukin (interleukin 2) and IFN-α2a therapy in metastatic renal cell carcinoma patients . Clin Cancer Res . 2009 ; 15 : 4986 – 4992 .
95. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function . Cancer Res . 2010 ; 70 : 5213 – 5219 .
96. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma . Clin Cancer Res . 2012 ; 18 : 1386 – 1394 .
97. Combining immunotherapy and targeted therapies in cancer treatment . Nat Rev Cancer . 2012 ; 12 : 237 – 251 .
98. Regulatory T-cells and associated pathways in metastatic renal cell carcinoma (mRCC) patients undergoing DC-vaccination and cytokine-therapy . PLoS One . 2012 ; 7 : e46600 .
99. Gene expression profile of peripheral blood lymphocytes from metastatic renal cell cancer patients treated with IL-2, interferon-alfa and dendritic cell vaccine . PLoS One . 2012 ; 7 : e50221 .
100. Phase III, randomized, open-label, multicenter trial (BREAK-3) comparing the BRAF kinase inhibitor dabrafenib (GSK2118436) with dacarbazine (DTIC) in patients with BRAFV600E-mutated melanoma . J Clin Oncol . 2012 ; 30 suppl;abstr lba8500 .
101. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAFV600E-mutated melanoma . J Clin Oncol . 2012 : 30 suppl;abstr 8502 .
102. Been there, not done that—melanoma in the age of molecular therapy . N Engl J Med . 2011 ; 364 : 2547 – 2548 .
103. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing . N Engl J Med . 2012 ; 366 : 883 – 892 .
104. The median isn’t the message . Discover . 1985 ; 6 : 40 – 42 .
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


