Despite vast phenotypic variation in the population, human beings are genetically very similar. One individual human genome differs from another by approximately 1 nucleotide in every 1,000 bp (0.1%). Thus, each haploid human genome of 3 × 109 bp harbors approximately 3,000,000 DNA variants, most of which are rare and predominantly found in noncoding DNA such as introns and intergenic regions.
All DNA Sequence Polymorphisms are Due to Mutations, but not All Mutations Result in Polymorphisms
A mutation is any change of an ancestral base-pair sequence to a different nucleotide. Rare DNA sequence variants may be unique to an individual or a family. In contrast, polymorphisms are common differences in DNA sequence, strictly defined as having a frequency ≥1% for the less common allele(s). Thus, all polymorphisms are due to mutations, but not all mutations result in polymorphisms.
A mutation may be (a) a single base-pair substitution, (b) a deletion or insertion of 1 or more base pairs (
indel), or (c) a larger deletion, insertion, or other rearrangement of genetic material. Mutations may be neutral (i.e., cause no observable change in phenotype or functional disruption) or deleterious (disease causing). In humans, new mutations occur approximately every 1 × 10
-8 bp/generation, which corresponds to approximately 60 new genetic mutations per individual.
However, >98.5% of mutations occur in introns or intergenic regions (noncoding regions), so it is difficult to assess the consequence and implication of these mutations for human health and disease susceptibility.
Most well-characterized disease-causing mutations occur in exons or in exon/intron junctions. DNA mutations may disrupt normal gene expression or function in several ways (See
FIGURE 3.1 and
Chapter 4).
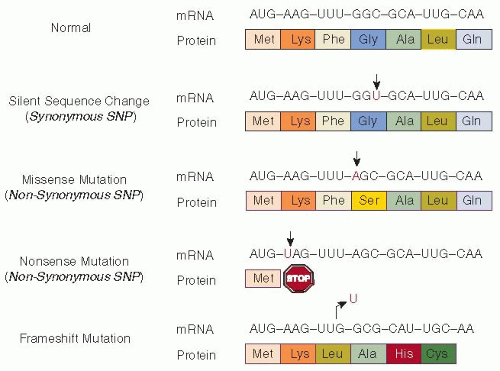
Point mutations are the replacement of a singlebase nucleotide with another nucleotide. Some point mutations within the coding sequence do not alter the encoded amino acid (
synonymous substitution) and have no effect on the function or expression of the gene. However, a subset of synonymous mutations may alter the structure, function, and expression level of proteins via other mechanisms.
7 Missense mutations (
nonsynonymous substitutions) are changes in the DNA sequence that cause one encoded amino acid to be changed to a different amino acid, thus changing the overall peptide sequence of the gene product. Some missense mutations are deleterious to protein function, while others may be neutral.
Nonsense mutations are another type of nonsynonymous substitution, changing a specific amino acid codon to a stop codon and thereby producing a shortened protein product. Frameshift mutations result from the insertion or deletion of a number of nucleotides (not divisible by 3) that alters the normal protein-coding reading frame. The reading frame of a DNA or RNA molecule refers to the sequence of three-letter codons that can be translated into amino acids. A frameshift mutation changes the amino acid sequence downstream of the mutation, often resulting in a premature stop codon. Of note, small insertions/deletions (indels) of a multiple of three nucleotides will insert or delete one or more amino acids, but are not frameshift mutations because they do not alter the reading frame.
Finally,
splice-site mutations result in an altered RNA sequence by changing the specific site at which splicing of an intron takes place during mRNA processing. Splice-site mutations may lead to the production of aberrant proteins. However, mutations far from a splice site may also affect splicing via alternate mechanisms
8 or may be implicated in abnormal polyadenylation of mRNAs, RNA stability, or transcript processing.
2 An important cellular mRNA surveillance mechanism known as
nonsensemediated decay (NMD) results in the instability of mRNAcontaining nonsense mutations, thereby reducing the expression of truncated, potentially harmful proteins.
9Mutations that functionally disrupt protein-coding genes, known as
loss-of-function (LOF) mutations, are surprisingly common in the human genome. LOF variants have historically been assumed to cause disease, as is the case for many Mendelian diseases. However, recent large-scale sequencing projects have revealed that apparently healthy individuals harbor at least 100 LOF variants in their genomes, including approximately 30 in the homozygous or compound heterozygous state.
10Some LOF variants that are common in healthy individuals have been known and well characterized for years, such as the O allele of the blood group ABO antigen locus, and other variants that contribute to variable drug-metabolizing capacity between individuals. Nevertheless, the finding that healthy individuals carry dozens to hundreds of seemingly benign LOF variants came as a great surprise to the scientific community. Even more startling is the mounting evidence that gene disruption may actually be beneficial in some cases.
10It is possible for one disease to result from many different types of mutations in a given gene. For example, over 1,000 unique mutations causing hemophilia A are reported in the worldwide hemophilia database (HAMSTeRS).
11 As a result of its X-linked inheritance, hemophilia A is observed with a high frequency in the human population (1:5,000 male newborns worldwide) (
Chapter 51). As Haldane predicted in 1935, one-third of males with a lethal X-linked disorder should represent
de novo somatic genetic mutations in eggs or sperm,
12 based on the assumption that one-third of lethal X-linked mutations are carried in males and will be lost in each generation. Thus, most hemophilia A mutations are expected to be on average only a few generations
old. Many of these mutations occur at CpG dinucleotides, which are known hotspots for mutation in mammalian genes.
13,14Mutations occurring outside of coding regions, such as in regulatory regions or in splice-site consensus sequences, may alter gene transcription or lead to alternative mRNA transcripts, respectively. Of historic interest, by analyzing DNA recovered from skeletal bone specimens of the Romanov family, a mutation in the
F9 gene predicted to alter RNA splicing was identified as the cause of the “Royal Disease” transmitted from Queen Victoria to the Royal families of Europe, thereby determining that this disease was hemophilia B, not the more common hemophilia A.
15Finally, in addition to smaller coding and noncoding mutations, structural rearrangements comprise another important class of genetic mutations, including deletions, insertions, inversions, and chromosomal translocations. Such larger genomic disruptions have been observed to cause hemophilia A, including Line 1 retrotransposon insertion.
11,16 Of particular note, the recurrent intron-22 inversion in the
FVIII gene accounts for approximately 35% to 45% of all severe hemophilia A cases.
17 Duplications or deletions of large segments of DNA termed
copy number variations (CNVs) are surprisingly common in the human genome. CNVs ranging in size from thousands to millions of base pairs may be present in anywhere from 0 to 2 or more copies when comparing one individual to the next.
18 CNVs can encompass entire genes or groups of genes, leading to gene dosage imbalance. Healthy individuals harbor multiple CNVs, which likely contribute to normal trait variation, though a subset of CNVs are associated with disease or disease susceptibility. Until recently, structural variants were technologically difficult to detect and characterize, as compared to single-nucleotide substitutions. Common structural variants are now estimated to involve between 9 to 25 Mb (0.5% to 1%) of the genome.
6
Genetic Polymorphisms
As defined above, the formal definition of a polymorphism is any sequence or trait for which the less common or minor form(s) exhibit a population frequency of ≥1% (
Table 3.1). These variants are considered to be
common in the population. Common variants are not generally deleterious or associated with disease, which is consistent with the expectation that deleterious mutations would not typically be expected to reach a frequency of ≥1% in the population.
Single-nucleotide polymorphisms (SNPs) represent positions in the genome where two or occasionally three alternative nucleotides are common in the population. Though the term “SNP” is typically used to refer to all single-nucleotide DNA variants, even those with minor allele frequencies ≤1%, the latter should more correctly be identified as SNVs. Unlike common polymorphisms with relatively high population frequencies, variants occurring at a frequency <0.01 in the population are considered to be rare. These variants are also known as “private” polymorphisms because they are often restricted to specific pedigrees in a population.
Of note, the terms
polymorphism and
neutral variant are often erroneously used synonymously. However, rare variants can be neutral and common polymorphisms can be deleterious (
Table 3.1). In clinical practice, rare sequence changes in a disease gene in a patient can often only be described as “unclassified variants” or “variants of unknown significance.” The relative frequency of neutral, near-neutral, and nonneutral genetic variants remains to be precisely determined (
Table 3.2).
19Balanced polymorphisms represent a special class of genetic variant observed under balancing selection. In this case, an individual who is heterozygous at a particular genetic locus has a greater fitness or survival advantage compared to either type of homozygous individual, a phenomenon known as
heterozygote advantage. For example, individuals with sickle cell trait (heterozygote carriers of the HbS mutation) are asymptomatic with normal life expectancy, though they are resistant to
Plasmodium falciparum malaria,
20 which is endemic in West Africa. A balance exists between selection against individuals who suffer from sickle cell anemia (homozygous for HbS) and selection for heterozygous HbS carriers who are resistant to malaria.