Molecular and Cellular Basis of Radiotherapy
15.1 INTRODUCTION
Since their discovery by Roentgen more than a century ago, x-rays have played a major role in modern medicine. The first recorded use of x-rays for the treatment of cancer occurred within 1 year of their discovery. Subsequently there has been intensive study of x-rays and other ionizing radiations, and their clinical application to cancer treatment has become increasingly sophisticated. This chapter and Chapter 16 review the biological effects of ionizing radiation and the application of that knowledge to cancer treatment.
The present chapter begins with a review of the physical properties of ionizing radiations, their interactions within the cell (membrane, cytoplasm, and nucleus), and the molecular and cellular processes that ensue. The effect of energy deposition in tissue is discussed with emphasis on the pathways that control cellular proliferation following exposure to ionizing radiation. Finally, various genetic and epigenetic factors known to influence the radiosensitivity of normal and tumor cells are described in the context of using molecularly-based targets for designing treatment and predicting response to radiotherapy.
15.2 INTERACTION OF RADIATION WITH MATTER
15.2.1 Types of Radiation, Energy Deposition, and Measurements of Radiation Dose
X- and γ-rays constitute part of the continuous spectrum of electromagnetic (EM) radiation that includes radio waves, heat, and visible and UV light (Fig. 15–1). All types of EM radiation can be considered as moving packets (quanta) of energy called photons. The amount of energy in each individual photon defines its position in the EM spectrum. For example, x-or γ-ray photons carry more energy than heat or light photons and are at the high-energy end of the EM spectrum. Individual photons of x-rays are sufficiently energetic that their interaction with matter can result in the complete displacement of an electron from its orbit around the nucleus of an atom. Such an atom (or molecule) is left with a net (positive) charge and is thus an ion; hence the term ionizing radiation. Typical binding energies for electrons in biological material are in the neighborhood of 10 eV (electron volts). Thus photons with energies greater than 10 eV are considered to be ionizing radiation, while photons with energies of 2 to 10 eV are in the UV range and are nonionizing. An interaction that transfers energy, but does not completely displace an electron, produces an “excited” atom or molecule and is called an excitation.
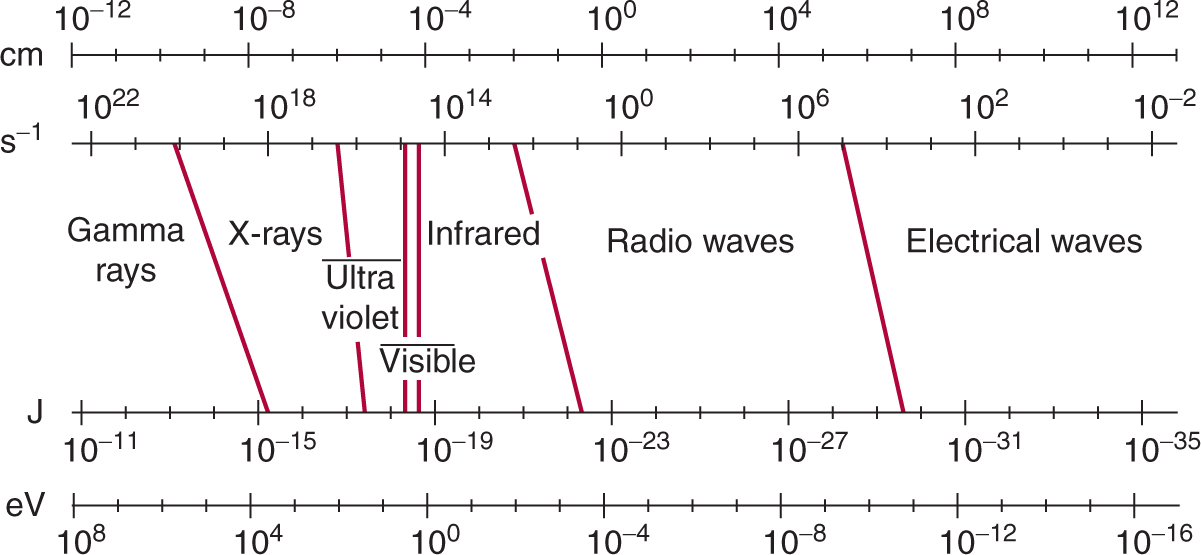
FIGURE 15–1 EM spectrum showing the relationship of photon wavelength in centimeters (cm) to its frequency in inverse seconds (s–1) and to its energy in joules (J) and electron volts (eV). The various bands in the spectrum are indicated. Slanted lines between bands indicate the degree of overlap in the definition of the various bands.
UV radiation is split into 3 general classes, UV-C, UV-B, and UV-A, corresponding to wavelengths of 200 to 290 nm, 290 to 320 nm and greater than 320 nm, respectively. UV-C and UV-B irradiation can be absorbed by DNA in cells leading to the production of various photoproducts, notably 6-4 photoproducts (6-4PPs) and interstrand crosslinks in the form of cyclobutane pyrimidine dimers. Such lesions may be repaired by a number of processes including nucleotide excision repair (see Chap. 5, Sec. 5.3.3). Because of the limited penetration of UV radiation through tissue, its effects in humans are primarily associated with the skin.
X-rays are produced when accelerated electrons hit a tungsten target and then decelerate emitting a spectrum of Bremsstrahlung radiation. The resulting spectrum is filtered to produce a clinically useful beam with minimal x-ray scatter from the axis of the central beam. When x-ray photons interact with tissue, they give up energy by 1 of 3 processes: the photoelectric effect, the Compton effect, or pair production. In the energy range most widely used in radiotherapy (100 keV to 25 MeV), the Compton effect is the most important mechanism leading to deposition of energy in tissue. This energy-transfer process involves a collision between the photon and an outer orbital electron of an atom, with partial transfer of energy to the electron and scattering of the photon into a new direction. The electron (and the photon) can then undergo further interactions, causing more ionizations and excitations, until its energy is dissipated. All 3 of the interaction processes mentioned above result in the production of energetic electrons that, in turn, lose energy by exciting and ionizing target atoms and molecules and setting more electrons in motion.
Modern clinical radiotherapy uses ionizing radiation to treat cancer patients and can be delivered to tissues by external beam radiotherapy using linear accelerators (ie, high-energy x-rays and electron beams), 60Co sources (γ-rays produced by radioactive decay as a result of unstable nuclei), or charged particle accelerators. Clinical radiotherapy can also be given using brachytherapy, which delivers highly localized radiation dose from within an organ or tissue using isotopes such as 131I, 125I, and 103Pd following radioactive decay (see Chap. 16, Sec. 16.2.3). The choice of the type of delivery of radiotherapy depends on tumor type and location within the body.
Because of their initial attenuation within the first few millimeters of tissue, low-energy x-ray beams (50 to 250 keV) deposit most of their energy close to the skin surface, and are therefore typically used to treat skin cancers or skin metastases. In contrast, high-energy x-ray beams (18 or 25 MeV) are less attenuated at the skin surface (ie, are “skin-sparing”) and deposit most of their energy at a greater depth within the body.
Charged particles can also be used for clinical radiotherapy and include electrons, light particles such as protons, and heavy particles such as carbon ions. Typically, electron beams are used to treat superficial tumors that require a uniform “skin to specific depth” dose distribution starting right from the outer skin surface while particle beams have been used for high-precision radiotherapy to treat tumors deep within the body because a monoenergetic particle beam has sharply defined edges and depth of penetration into tissue. Neutrons have also been used in radiotherapy. They have no charge and deposit energy by collision with nuclei, particularly hydrogen nuclei (protons), thereby transferring their energy to create moving charged particles capable of both ionization and excitation. In contrast to electromagnetic (EM) forms of radiotherapy in which the radiotherapy dose falls off exponentially over the depth of tissue treated, particle-based radiotherapy deposits its energy at a depth within a Bragg peak (see Fig. 15–2 and Chap. 16, Fig. 16–3A).
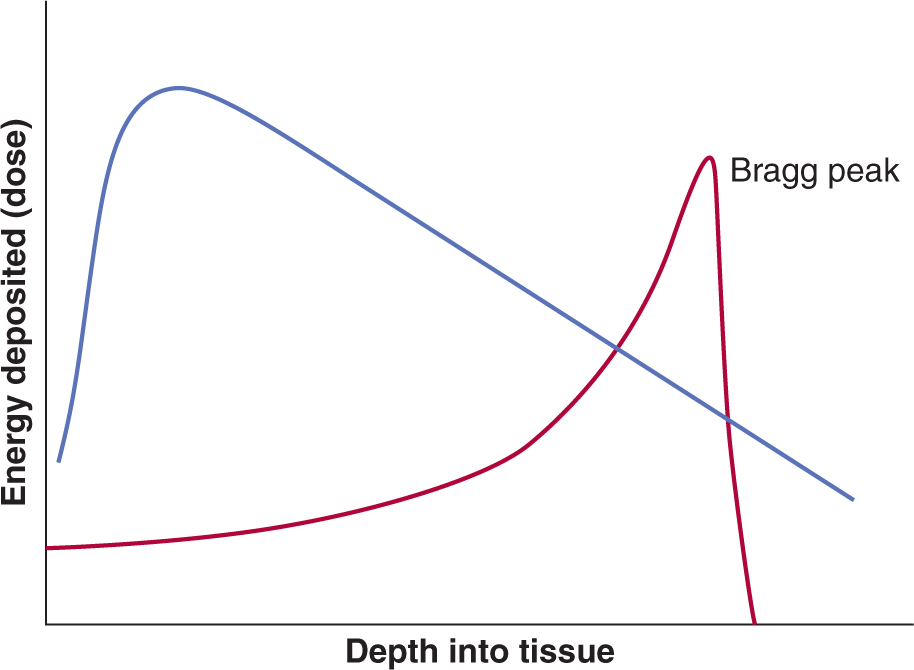
FIGURE 15–2 Schematic of the energy deposition by electromagnetic (EM) photon radiotherapy (pink curve) versus a charged particle along its track in tissue (red curve). The particle has a high velocity at the left-hand side of the figure, but as it loses energy, it slows down until it comes to rest in the region of the Bragg peak. In contrast, the electromagnetic (EM) form of photon radiotherapy leads to absorbed dose that falls off exponentially over the depth of tissue treated.
Radiation dose is measured in terms of the amount of energy (joules) absorbed per unit mass (kg) and is quoted in grays (1 Gy is equivalent to 1 J/kg). It is not the total amount of energy absorbed that is critical for the biological effect of ionizing radiation. For example, a whole-body dose of 8 Gy would result in the death (caused by bone marrow failure) of many animals, including humans, yet the amount of energy deposited, if evenly distributed, would cause a temperature rise of only about 2 × 10–3 °C. It is the size and localized nature of the individual energy-deposition events caused by ionizing radiations that is the reason for their efficacy in damaging biological systems.
15.2.2 Linear Energy Transfer and Energy Absorption
The deposition of energy in matter by moving charged particles is chiefly a result of electrical field interactions and depends both on the velocity and on the charge of the particle. As a charged particle moves through matter, it transfers energy by a series of interactions that occur at random. The particle’s energy loss dE, along a portion of its track dx, is dependent on its velocity v, charge Z, and the electron density of the target ρ as indicated by mathematical relationship as Equation 15.1:
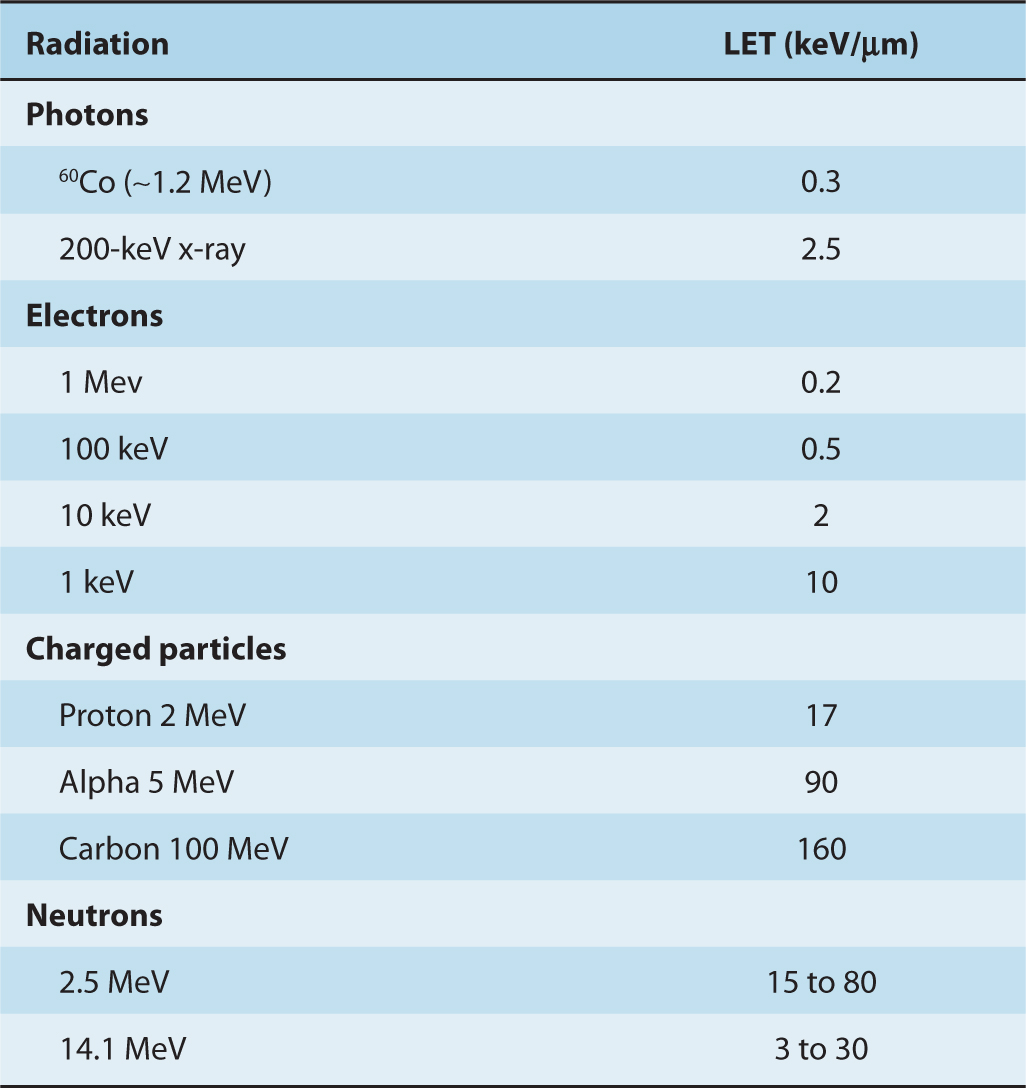
The efficiency with which different types of ionizing radiation cause biological damage varies, even though photons, charged particles, and neutrons ultimately all set electrons in motion. The important difference between different types of radiation is the average density of energy loss along the track of the particle. The average energy lost by a particle over a given track length is known as the linear energy transfer (LET). The units of LET are given in terms of energy lost per unit path-length, for example, keV/μm. Table 15–1 provides some representative values of LET for different particles. Because the energy of the particle depends on its mass (m) and velocity (E = mv2), it can be seen from Equation 15.1 that as a particle slows down, it loses energy more and more rapidly and reaches a maximum rate of energy loss (the Bragg peak) just before it comes to rest (Fig. 15–2). The LET of an individual charged particle thus varies along the length of its track.
TABLE 15–1 Linear energy transfer of various radiations.
The biological effect of a dose of radiation depends on its LET; it is therefore necessary to know the LET at each point in an irradiated volume to predict the biological response accurately. When EM radiation (eg, low-LET 6-MV photons, produced by a linear accelerator) is used to irradiate tissue, electrons are set in motion in the tissue. Because of their small mass (1/1860 of the mass of a proton), they are easily deflected and their track through the tissue is tortuous. Each electron track has a Bragg peak at its termination and a range of LET values along its track, but both the initiation and termination points of the electron tracks occur at random in the tissue, so that the LET spectrum is similar at all depths. A similar result occurs if the irradiation is with a primary electron beam. In contrast, if a beam of monoenergetic heavier-charged particles (eg, high-energy protons or carbon nuclei) is used to irradiate the tissue, the tracks of the particles are much straighter because their much larger mass (than the electrons in the tissue with which they mostly interact) reduces the chance of significant deflection. The Bragg peak then occurs at a similar depth in the tissue for all particles. Thus, there is a region in the tissue where a relatively large amount of energy is deposited. This feature of irradiation with high-LET particles makes them potentially attractive because the beam can be designed to deposit most of its energy in a deep-seated tumor (see Chap. 16, Sec. 16.2.4).
15.2.3 Radiation Damage within the Cell
The interactions leading to energy deposition in tissue occur very rapidly and generate chemically reactive free electrons and free radicals (molecules with unpaired electrons). Many different molecules in cells are altered either as a result of direct energy absorption or as a result of energy transfer from one molecule to another, giving rise to indirect effects (Fig. 15–3). Most of the energy deposited in cells is absorbed initially in water (because the cell is approximately 80% water), leading to the rapid (ie, within 10–14 to 10–4 s) production of reactive intermediates (oxidizing and reducing radicals), which, in turn, can interact with other molecules in the cell (indirect effect). The hydroxyl [OH˙] radical, an oxidizing agent, is probably the most damaging. The cell contains naturally occurring thiol compounds such as glutathione, cysteine, cysteamine, and metallothionein, whose sulfhydryl (SH) groups can react chemically with the free radicals to decrease their damaging effects. Other antioxidants include the vitamins C and E and intracellular manganese superoxide dismutase (MnSOD). The intracellular levels of thiols and antioxidative molecules may differ between normal and tumor tissues, and their manipulation may offer a clinical strategy to protect normal tissues from radiotherapy-induced damage. One example is the use of the thiol-containing drug, amifostine to protect against radiotherapy-induced xerostomia (ie, dry mouth) after irradiation of salivary glands (see Chap. 16, Sec. 16.5.7).
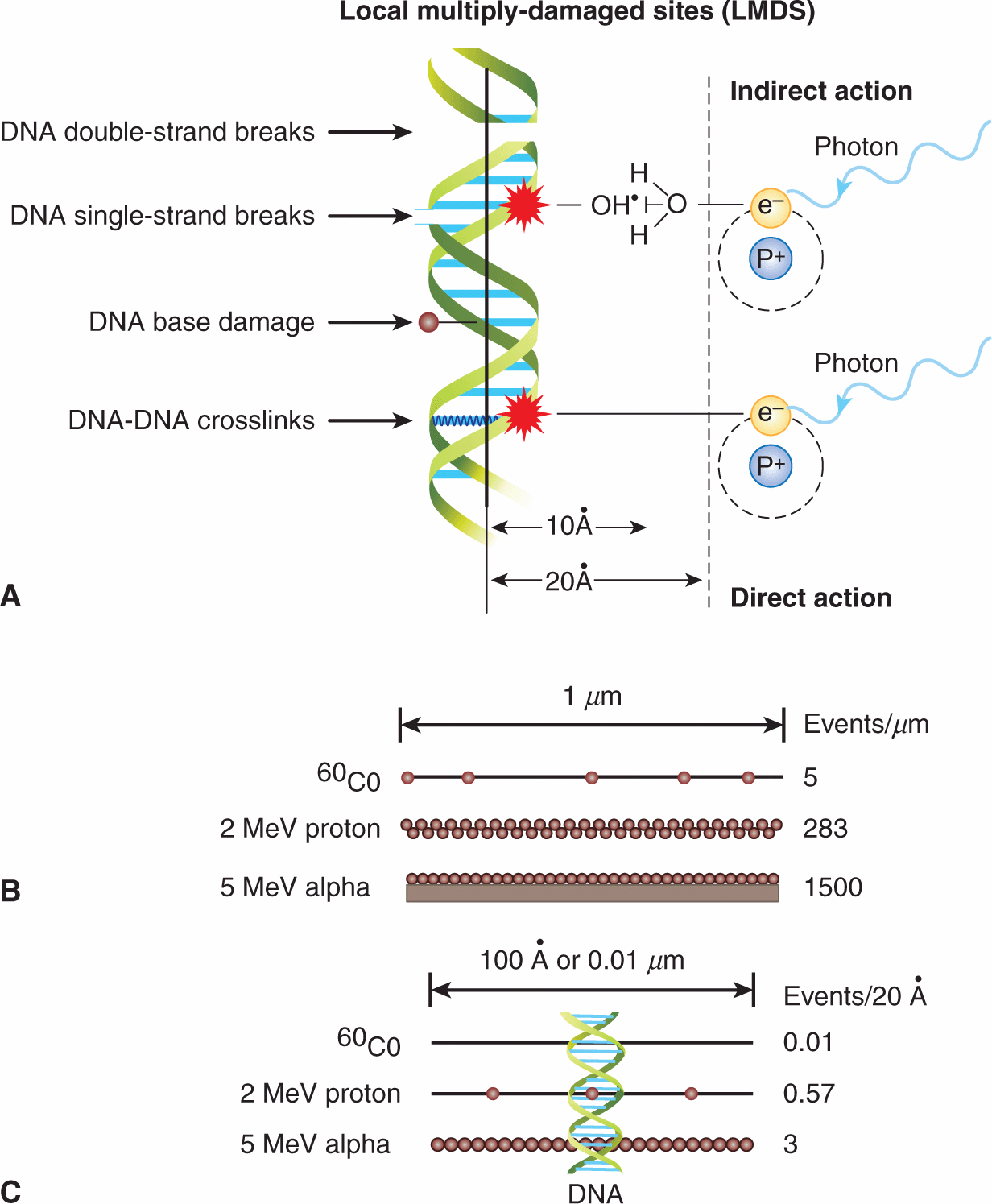
FIGURE 15–3 A) Direct and indirect effects of ionizing radiation on DNA. In the indirect model, chemically reactive free radicals are produced during ionization of water molecules in close proximity (ie, 10 to 20 angstroms [Ã]) to the DNA helix. These free radicals, such as the hydroxyl radical, OH’, can react chemically with DNA to produce DNA damage. In the direct model, absorption of the energetic electron occurs directly within the DNA causing localized damage without an intermediate free radical step. Indirect and direct damage can lead to clusters of DNA single-strand and double-strand breaks, DNA base damage, DNA–DNA or DNA–protein crosslinks. These can occur as local multiply-damaged sites (LMDS). (Adapted from Hall, 2000.) B) and (C) The frequency of primary energy-loss events along the tracks of various radiations of widely differing linear energy transfer. B) Schematic of primary energy-loss events over a distance of 1 μm. C) Primary energy-loss events over 0.01 μm or 100 Ã, depending on type of radiation. The dimensions of a DNA double helix are illustrated.
The random nature of the energy-deposition events means that radiation-induced changes can occur in any molecule in a cell. DNA is a major target of ionizing radiation, because of its biological importance to the cell. Even relatively small amounts of DNA damage can lead to cell lethality. It has been estimated that approximately 105 ionizations can occur within a diploid cell per gray of absorbed radiation dose, leading to approximately 1000 to 3000 DNA–DNA or DNA–protein crosslinks, 1000 damaged DNA bases, 500 to 1000 single-strand and 25 to 50 double-strand DNA breaks (ie, the vast majority of the ionization events do not cause DNA damage). Focal areas of DNA damage can arise because of the clustering of ionizations within a few nanometers of the DNA (Ward, 1994). These “local multiply damaged sites” (LMDS; see Fig. 15–3) include combinations of single- or double-strand breaks in the sugar-phosphate backbone of the molecule, alteration or loss of DNA bases, and formation of crosslinks (between the DNA strands or between DNA and chromosomal proteins). Most DNA lesions can be repaired by DNA repair pathways (see Chap. 5, Sec. 5.3) probably acting together to repair clustered LMDS-associated lesions. High LET irradiation causes an increase in both the number and complexity of DNA clustered lesions and is more difficult to repair.
Most studies suggest that cell death following radiation is correlated with either initial or residual levels of DNA double-strand breaks (Jeggo and Lavin, 2009). Table 15–2 outlines evidence to support the role of damage to DNA as a crucial type of cellular damage in relation to cell killing.
TABLE 15–2 Evidence supporting DNA as a critical target for radiation-induced lethality.
The reactive oxygen species (ROS) induced by ionizing radiation can also interact with proteins in the cell membrane, some of which may be involved in signal transduction. This can lead to apoptosis in certain cell types (eg, endothelial cells) by activation in the membrane of a ceramide-sphingomyelin pathway (see Sec. 15.3.2; Fuks et al, 1995). Furthermore, preincubation of cells with agents capable of altering either protein function or lipid peroxidation within the cell membrane, including anti-ceramide antibodies, can also modify the level of radiation-induced apoptosis (Fuks et al, 1995; Rotolo et al, 2012). Overall, the cellular response to ionizing radiation is mediated both by the direct damage to DNA and a complex interaction between proteins located within the plasma membrane, cytoplasm, and nucleus of the cell (see Sec. 15.3.2).
15.2.4 Genetic Instability, Chromosomal Damage, and Bystander Effects
Many human cancers contain chromosomal rearrangements, including chromosomal translocations, deletions, and amplifications. Chromosome rearrangements can also be observed in cells after irradiation, and if nonlethal, may contribute to the carcinogenic properties of ionizing radiation (see Chap. 16, Sec. 16.8). Chromosomal instability following irradiation has been demonstrated using a variety of techniques including spectral karyotyping (SKY) and fluorescence in situ hybridization (FISH) (see Chap. 2, Sec. 2.2.6). DNA double-strand breaks can lead to chromosomal rearrangements at the first mitosis after exposure to ionizing radiation and the type of aberration (ie, chromosomal versus chromatid types of rearrangements; Fig. 15–4) reflects the cell-cycle phase at the time of irradiation. Pathways for rejoining of DNA double-strand breaks in mammalian cells, include homologous recombination and nonhomologous end-joining (see Chap. 5, Sec. 5.3), but it is unclear which factors determine whether or not an induced break will lead to a chromosomal rearrangement.
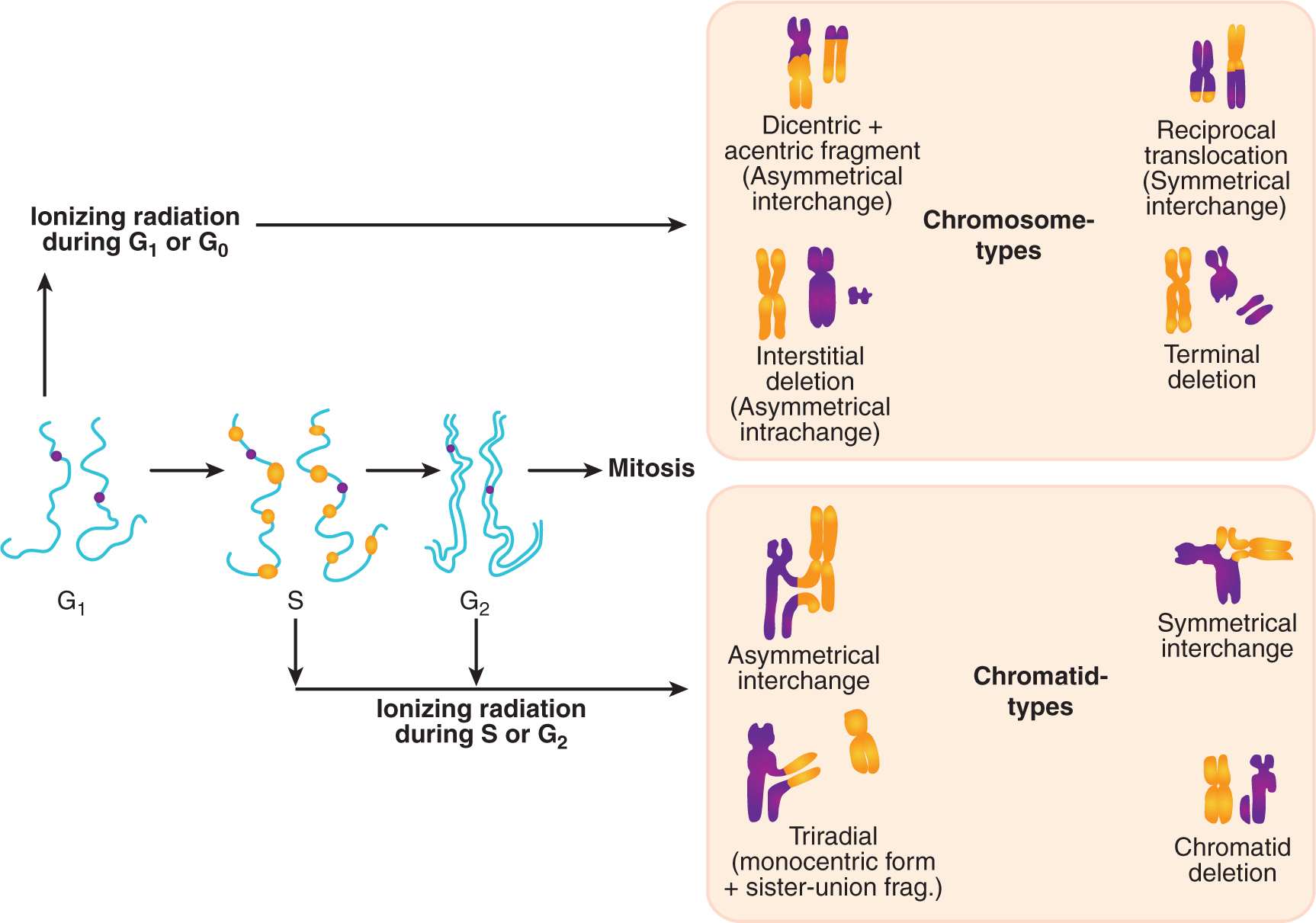
FIGURE 15–4 Chromosomal versus chromatid types of radiation-induced damage as a function of cell-cycle phase during irradiation. If a cell is irradiated during the G1 or early S phase when only 1 chromosomal homolog exists, a nonrepaired DNA-DSB (double-strand break) can lead to dicentrics, reciprocal translocations and acentric fragments. During irradiation in late S and G2 phases, sister chromatids have duplicated and the DNA-DSB gives rise to interchanges, triradials, or a chromatid deletion. Consequently, the type of chromosomal damage observed may also reflect the relative ability of cells to utilize the nonhomologous end-joining (optimally used during G1) or homologous recombination (optimal in S and G2) repair pathways to correct the DNA-DSB. (From Nagasawa et al, 2010.)
Examination of the break points in very large deletions often show that novel chromosomal fragments found in rearrangements are derived from other chromosomal sites of radiation damage in the same cell; they can arise from complex interactions between a number of damaged sites in the cellular DNA (Singleton et al, 2002). These rearrangements occur preferentially between genomic regions that are located in a similar vicinity in the nucleus (Misteli, 2009). Furthermore, if a cell survives and proliferates after irradiation, delayed chromosomal instability can be observed in the descendants of the exposed cell. Such radiation-induced chromosomal instability may be secondary to a breakage-fusion-bridge cycle (see Chap. 5, Sec. 5.7) or to epigenetic effects which perpetuate the unstable phenotype in irradiated cells (Huang et al, 2003).
Reactive oxygen species (ROS) or other factors may be released from an irradiated cell to cause damage to neighboring nonirradiated cells (ie, bystander cells). Thus, media transfer from irradiated cells to an unirradiated cell population can lead to cell death in the nonirradiated cells. Similarly, targeting of 10% to 30% of a cell population with high LET irradiation using a focused microbeam can lead to cell death in the non-targeted surrounding cells within the culture dish (Fig. 15–5). The mechanisms for these nontargeted effects of radiation and their importance for radiation effects in vivo remain uncertain but appear to involve stress signaling pathways. This is a developing area of radiobiological study, particularly since bystander effects of radiation have implications for assessment of radiation effects in multicellular organisms and for health risks associated with radiation exposure (Morgan and Sowa, 2009; Blyth and Sykes, 2011; Mothersill and Seymour, 2012).
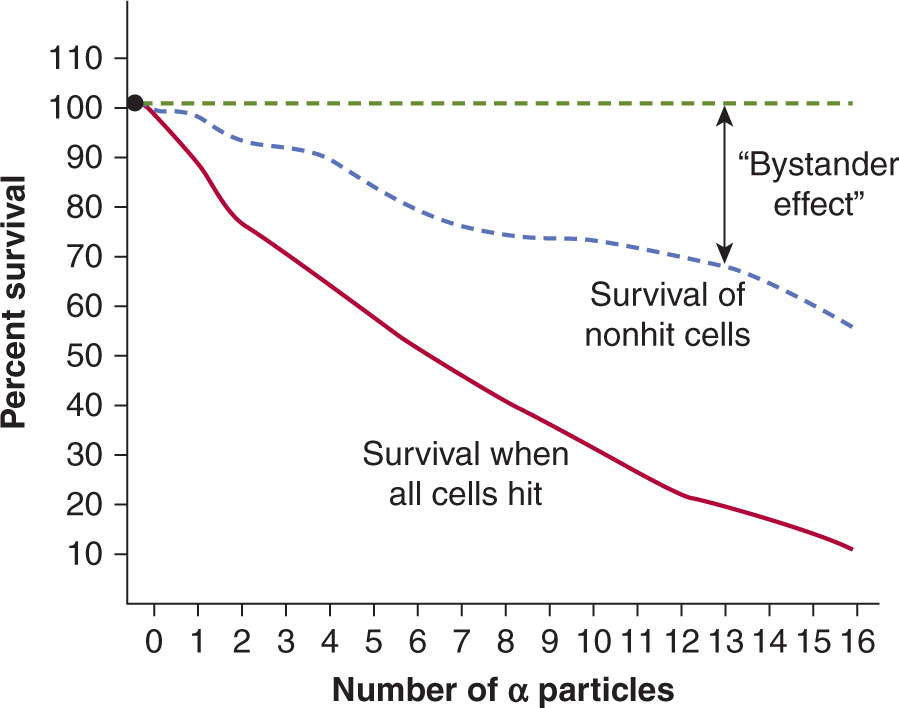
FIGURE 15–5 The radiation bystander effect. Direct single-cell irradiation of V79 cells with α particles can be accomplished using a specialized targeting microbeam in which the irradiation and fate of single cells can be tracked postirradiation. It was observed that cells that were not targeted by the α particles can also be killed (ie, they do not form colonies). This radiation bystander effect may be secondary to factors released by Irradiated cells into the surrounding media. In the plot shown, direct cell kill increases with increasing α-particle dose following single-cell irradiation. However, the death of nonirradiated cells also increases as a function of dose. The difference between an expected survival of 100% for nonirradiated cells and the actual survival observed, reflects the extent of cell kill by the bystander effect. (Adapted from Hall and Hei, 2003.)
15.3 CELL DEATH RESPONSES TO IONIZING RADIATION
15.3.1 In Vitro and In Vivo Assays for Radiation Cell Survival
Inhibition of the continued reproductive ability of cells is an important consequence of the molecular and cellular responses to radiation, as it occurs at relatively low doses (a few grays) and it is the major aim of clinical radiotherapy. A tumor is controlled if its stem cells are prevented from continued proliferation. A cell that retains unlimited proliferative capacity after radiation treatment is regarded as having survived the treatment. In contrast, a cell that has lost the ability to generate a “clone” or colony is regarded as having been killed, even though it may undergo a few divisions or remain intact for a substantial period before it lyses and is lost from the cell population. Colony formation following irradiation is thus an important end point, because it relates to a cell’s ability to repopulate normal or tumor tissues following exposure to ionizing radiation (see also Chap. 17, Sec. 17.4.1). In the assay that is used most often to assess colony formation, cells grown in culture are irradiated either before or after preparation of a suspension of single cells and plated at low concentration in tissue-culture dishes. Following irradiation, the cells are incubated for a number of days, and those that retain proliferative capacity divide repeatedly to form discrete colonies of cells (Fig. 15–6). After incubation, the colonies are fixed and stained so that they can be counted.
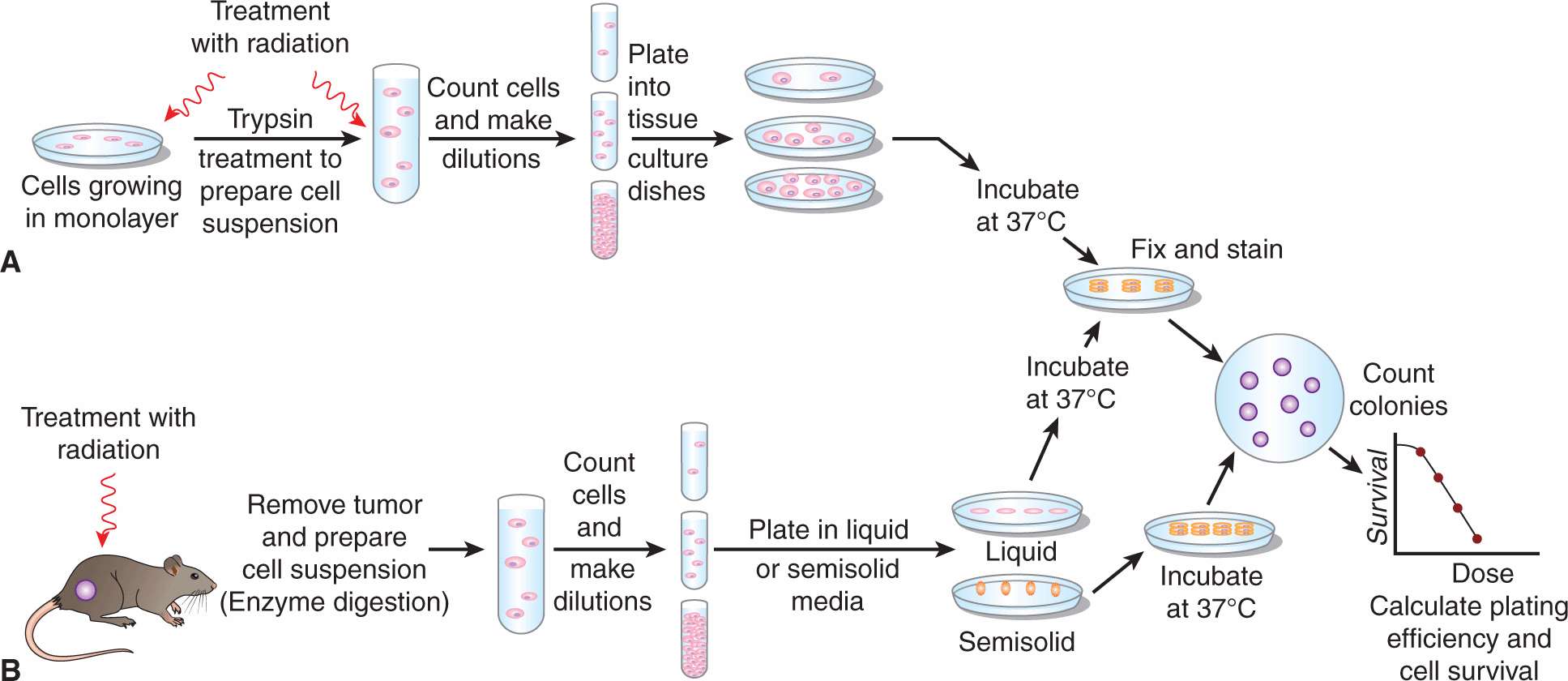
FIGURE 15–6 Schematic of in vitro plating assays to assess cell survival. A) Assay for the radiation sensitivity of cells growing in culture. B) In vivo–in vitro assay for the sensitivity of tumor cells grown and irradiated in vivo.
Cells that do not retain proliferative capacity following irradiation (ie, are killed) may divide a few times, but form only very small “abortive” colonies. If a colony contains more than 50 cells (ie, derived from a single cell by at least 6 division cycles), it is usually capable of continued growth and can be regarded as having arisen from a surviving cell. The plating efficiency (PE) of the cell population is calculated by dividing the number of colonies formed by the number of cells plated. The ratio of the PE for the irradiated cells to the PE for control cells is calculated to give the fraction of cells surviving the treatment (cell survival/surviving fraction). If a range of radiation doses is used, then these cell-survival values can be plotted to give a survival curve, such as the ones shown in Figures 15–7 and 15–8. Cells taken directly from animal or human tumors can also be grown in culture, allowing the in vitro assay to be extended to study the radiation sensitivity of tumor cells treated in vivo (see Fig. 15–6). Untreated cells rarely have a PE of 1 (more usually it is 0.5 to 0.8 for cells passaged for many generations and much lower for cells derived from spontaneous tumors).
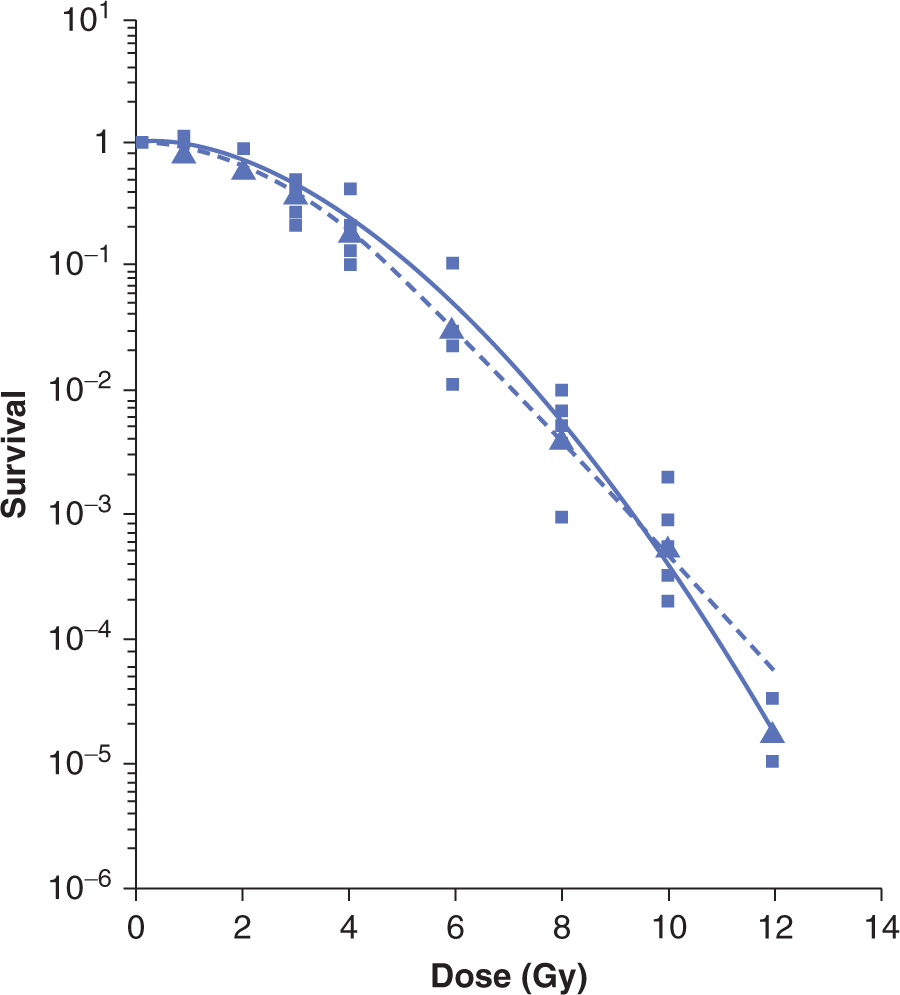
FIGURE 15–7 Survival data for a murine melanoma cell line treated with low-LET (γ-rays) radiation. The survival is plotted on a logarithmic scale against dose plotted on a linear scale. The data from 5 independent survival experiments are shown as the small squares, with the geometric mean value at each dose shown as the large triangles. The survival curves shown are the result of fitting the data to target theory (dashed line) or linear quadratic (solid line) models (Appendix 15.1). (Adapted from Bristow et al, 1990.)
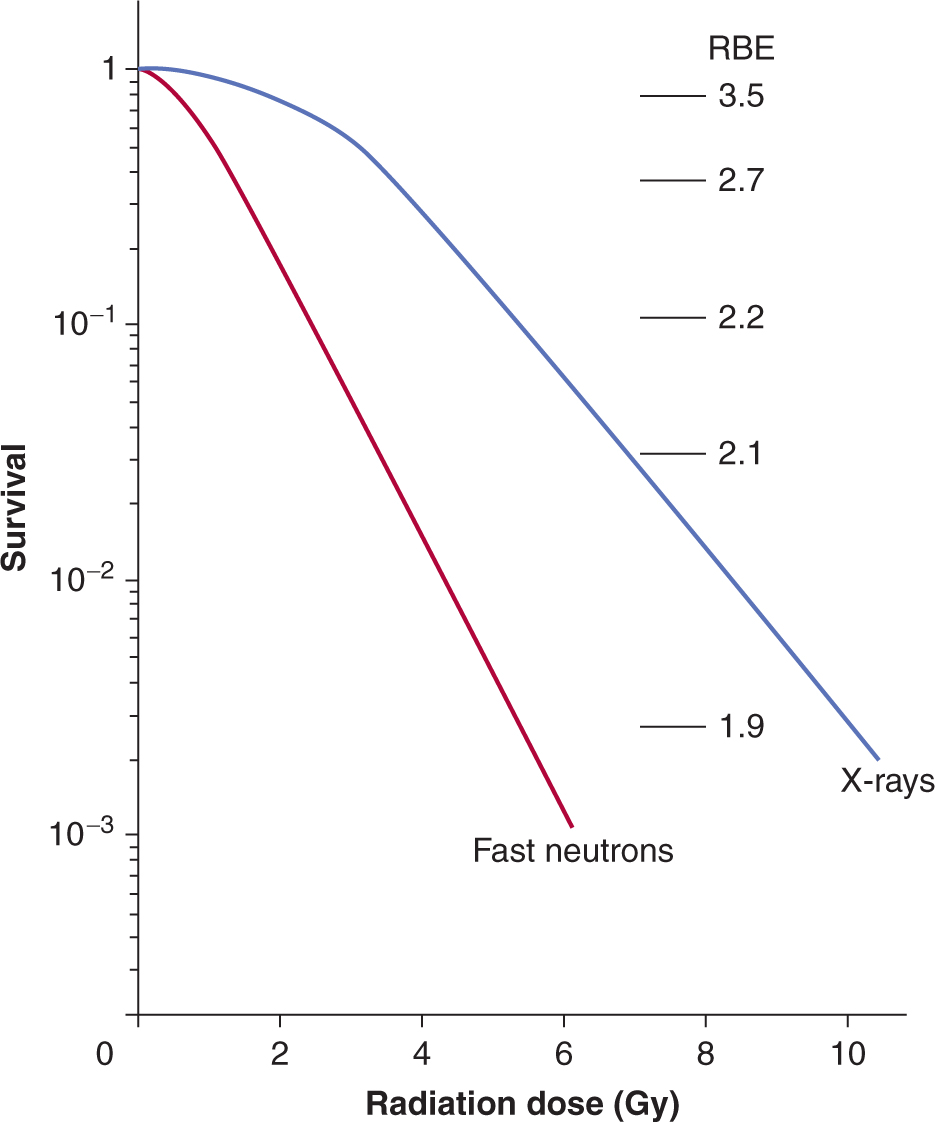
FIGURE 15–8 Comparison of survival curves for low-LET (x-ray) and high-LET (fast-neutron) radiation. The RBE (relative biological effectiveness) is calculated as indicated in the text and varies at different levels of survival.
The techniques described above have been used to obtain survival curves for a wide range of malignant and normal cell populations. In general, for low-LET radiation (eg, X-or γ-rays), these curves have the shape(s) illustrated in Figure 15–7, when cell survival is plotted on a log scale as a function of dose plotted on a linear scale. At low doses, there is evidence of a shoulder region; but at higher doses, the curve either becomes steeper and straight so that survival decreases exponentially with dose (dotted line in Fig. 15–7) or appears to be continually bending downward on the semilogarithmic plot (solid line in Fig. 15–7). There is greater variation in the low-dose or shoulder region of the radiation survival curves obtained for mammalian cells as compared to the variation in the slopes of the high-dose region of the curves (see Chap. 16, Sec. 16.3.2).
Figure 15–8 illustrates the difference in survival curves for X- or γ-rays (low-LET) and for fast-neutron (high-LET) irradiation. In general, the shoulder of the survival curve is reduced for higher-LET radiation and the curve is steeper. The biological effectiveness (RBE) of different types of radiation can be defined as the ratio of the dose of a standard type of radiation to that of the test radiation that gives the same biological effect. The standard type of radiation was originally taken as 200- or 250-kVp x-rays. Cobalt-60 γ-rays are also used as a standard for comparison studies, although their RBE relative to 250-kVp x-rays is approximately 0.9. Because the shoulder of the survival curve is reduced for high-LET radiation, the RBE varies with the dose or the survival level at which it is determined (see Fig. 15–8).
Many different mathematical models have been used to produce equations that can fit survival-curves to data within the limits of experimental error. Two commonly used models are the target-theory and linear-quadratic models of cell survival (explained in detail in Appendix 15.1) from which parameters (D0, n, or α and β respectively) can be used to describe the shape of the low-dose and high-dose regions of mammalian clonogenic cell survival curve. The accuracy of the data is usually such that either shape could fit the data adequately over the first few decades of survival, as illustrated in Figure 15–7. However, such descriptions are useful when comparing cellular radiosensitivity amongst a variety of cell types or when the shape of the survival curve is altered following treatment with drugs or changes in the environment (eg, hypoxia; see Chap. 16, Sec. 16.4.1).
Nonclonogenic assays have also been used to estimate the relative radiosensitivity of cells, although assays that measure short-term growth or programmed cell death/apoptosis (see Chap. 17, Sec. 17.4.1 and Sec. 15.3.2) often do not correlate with colony-forming assays. Assays for apoptosis may predict clonogenic survival for lymphoma and testicular cancer cell lines, as these cell types tend to die uniformly by apoptosis following irradiation. Assays that evaluate cellular growth for a short period (eg, 1 to 5 days) following radiation, such as the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) (or similar) assay that determines cellular viability by colorimetric assessment of the reduction of a tetrazolium compound, are also useful primarily for cells that die by apoptosis. They are of limited value for radiosensitivity studies of other cell types because they usually do not reflect the later death (following a few cell divisions) that is detected with a colony-forming assay. Such assays also have a limited range and it is rarely possible to assess more than one decade of cell kill. At present, clonogenic survival remains the “gold standard” for determining the radiosensitivity of cells in vitro.
Methods have been developed for assessing the ability of cells to form colonies in vivo. One of these is the spleen-colony method, which has been used to assess both the radiation and drug sensitivity of specific bone marrow (stem) cells (McCulloch and Till, 1962). In this assay, bone marrow from treated animals is injected into irradiated new hosts and colonies from surviving bone marrow (spleen colony-forming) cells can be then counted in the spleen. Initially these cells were thought to be hematopoietic stem cells but it is now known that they are downstream of the true hematopoietic stem cells in the maturation hierarchy (see Chap. 17, Fig. 17–10). In an analogous method, the lung-colony assay, tumor cells from treated animals are injected intravenously and form colonies in the lungs of syngeneic mice (Hill and Bush, 1969). Other colony-forming assays have been developed to study the radiation response of stem cells in situ in certain proliferative tissues, including skin, and gastrointestinal tract (see Chap. 16, Sec. 16.5.1).
15.3.2 Radiation-Induced Cell Death: Apoptosis, Autophagy, Mitotic Catastrophe, and Terminal Growth Arrest
Table 15–3 outlines mechanisms of cell death following irradiation. Many types of cells do not show morphological evidence of radiation damage until they attempt to divide. The morphology of the cell at the time of cell lysis following irradiation can be apoptotic or necrotic (see Chap. 9, Sec. 9.4.1). Following doses of less than approximately 10 Gy, lethally damaged cells may: (a) undergo a permanent (terminal) growth arrest, (b) undergo interphase death and lysis as a result of radiation-induced apoptosis or necrosis, or (c) undergo up to 4 abortive mitotic cycles and then finally undergo cell lysis as a result of mitotic catastrophe. The process of autophagy, which is primarily a cell-survival mechanism (see Chap. 12, Sec. 12.3.7), may also contribute to overall cell death within an irradiated population (Chaachouay et al, 2011; Anbalagan et al, 2012; Speirs et al, 2012).
TABLE 15–3 Characteristics of different modes of cell death following cellular exposure to ionizing radiation.
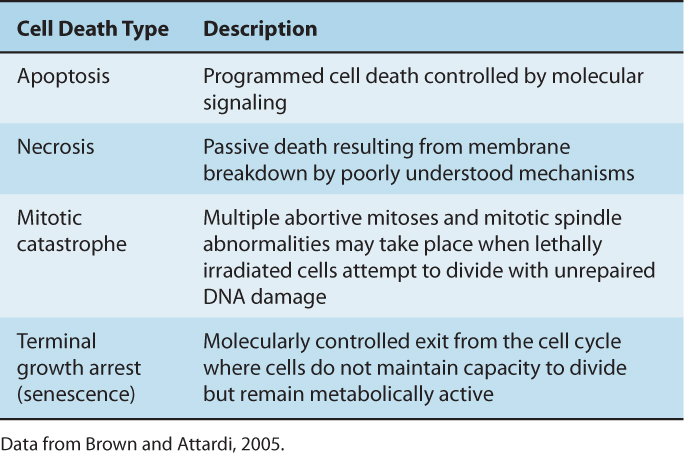
A radiation survival curve based on a clonogenic assay represents the total or cumulative cell death within an irradiated cell population as a result of all types of cell death. The biochemical and morphological differences observed for cells undergoing these processes are reviewed in Chapters 9 (Sec. 9.4) and 12 (Sec. 12.3.7). They can be related to loss of reproductive integrity defined by a colony-forming assay to determine the dominant mode of radiation-induced cell death within a given cell type.
For the majority of normal and tumor cells, death secondary to mitotic catastrophe accounts for most of the cell kill following irradiation. However, in some radiosensitive cells and the cancers that arise from them—notably lymphocytes, spermatocytes, thymocytes, and salivary gland epithelium—irradiation causes the cells to undergo an early (within a few hours) interphase death. This death is associated with the biochemical and morphological characteristics of apoptosis (ie, cell membrane blebbing, the formation of nuclear apoptotic bodies, and specific DNA fragmentation patterns). Depending on the type of cell, the intracellular target(s) for the induction of the apoptotic response may be either the cell membrane or the DNA or both. The reasons why some cell types undergo extensive radiation-induced apoptosis within a few hours after irradiation, while others do not, remains uncertain but may relate to the relative expression and function of proteins which trigger an apoptotic response. For example, in hematopoietic cells, radiation can lead to upregulation of genes (such as fas, bax, and caspase-3), which can facilitate apoptosis and/or downregulation of genes (such as bcl-2), which act to prevent apoptosis (Kitada et al, 1996).
Ionizing radiation can also initiate a sphingomyelin-dependent signaling pathway within the cell membrane, which can induce apoptosis in the absence of DNA damage (Ruiter et al, 1999; Kolesnick and Fuks, 2003). Ceramide is generated from sphingomyelin (SM) by the action of acid sphingomyelinase (ASM), or by de novo synthesis coordinated through the enzyme ceramide synthase. In endothelial, lymphoid, and hematopoietic cells, ceramide mediates apoptosis, whereas in other cells, ceramide may play no role in the death response. The ceramide-mediated apoptotic response to radiation can be inhibited by basic fibroblast growth factor (bFGF) or by genetic mutation of ASM. Radiation-induced crypt damage, organ failure, and death from the GI syndrome were reduced when apoptosis of supporting vascular endothelial cells was inhibited by intravenous bFGF or by deletion of the ASM gene (Paris et al, 2001). In animal studies, sphingosine 1-phosphate preserved fertility in irradiated female mice without propagating genomic damage in offspring, suggesting it might have a role to reduce radiotherapy-induced sterility during clinical treatment (Paris et al, 2002).
Some tumors may evade radiation-induced apoptosis by carrying p53 gene mutations or by lacking p53 expression or function, suggesting that restoration of wild-type p53 function using gene therapy might potentiate radiation cell kill (Ma et al, 2003). However, the level of radiation-induced apoptosis does not correlate with cell survival as measured by colony-forming assays for most types of tumor, so this approach is likely to have limited application (Brown and Wouters, 2001).
Autophagy is an important process responsible for degrading and recycling long-lived proteins, cellular aggregates and damaged organelles. A number of cellular stressors can activate autophagy in tumor cells leading to the process of selective cellular self-consumption (see Chap. 12, Sec. 12.3.7). One such stress is hypoxia (see Chap. 16, Sec. 16.4.1) and autophagy-mediated survival of hypoxic cells may affect tumor response to irradiation (Kim et al, 2009; White and DiPaola, 2009). Inhibitors of autophagy might be useful in augmenting radiation-induced cell death (Kim et al, 2009).
Most tumor cell lines retain the capacity of normal cells to undergo accelerated senescence after irradiation, and although the p53 and p21 genes act as positive regulators of treatment-induced senescence, they are not required for this response in tumor cells (Chang et al, 2000). Senescent or terminally-arrested cells are metabolically active but do not proliferate and do not form colonies following irradiation; they eventually undergo necrosis days to weeks following irradiation (ie, they are already dead in the clonogenic sense). This may explain the relatively slow resolution, yet ultimate cure, of some tumors following radiotherapy. Treatments that differentially increase terminal arrest in tumor cells might sensitize tumors to radiotherapy. For example, differentiation agents (eg, retinoids) have been used to induce a senescence-like phenotype and can radiosensitize both breast cancer and head and neck cancer cells in vitro and in vivo (Ma et al, 2003).
15.4 MOLECULAR AND CELLULAR RESPONSES TO IONIZING RADIATION
15.4.1 Cell-Cycle Sensitivity and DNA Damage Checkpoints
Mammalian cells respond to ionizing radiation by delaying their progression through the cell cycle. Such delays allow for the repair of DNA damage in cells prior to undergoing either DNA replication or mitosis and are thought to prevent genetic instability in future cell generations (Kastan and Bartek, 2004). There is a rapid decrease in the mitotic index in an irradiated cell population, as both lethally damaged and surviving cells ceased to enter mitosis, while cells already in mitosis continued to progress to the G1 phase. After a period of time, which depends on both the cell type and the radiation dose, surviving cells reenter mitosis (Fig. 15–9); this time is known as the mitotic delay. Mitotic delay appears to be due largely to a block of cell-cycle progression in G2 phase, although cells in G1 and S phases are also delayed to a lesser extent in their progression through the cell cycle. There is typically 3 to 4 hours of G2 delay per 1 Gy radiation in diploid cells. Cells may continue to experience delays in their progression through the next and subsequent cell cycles. As a result of radiation-induced delays in the cell cycle, cell populations can be partially synchronized by irradiation.
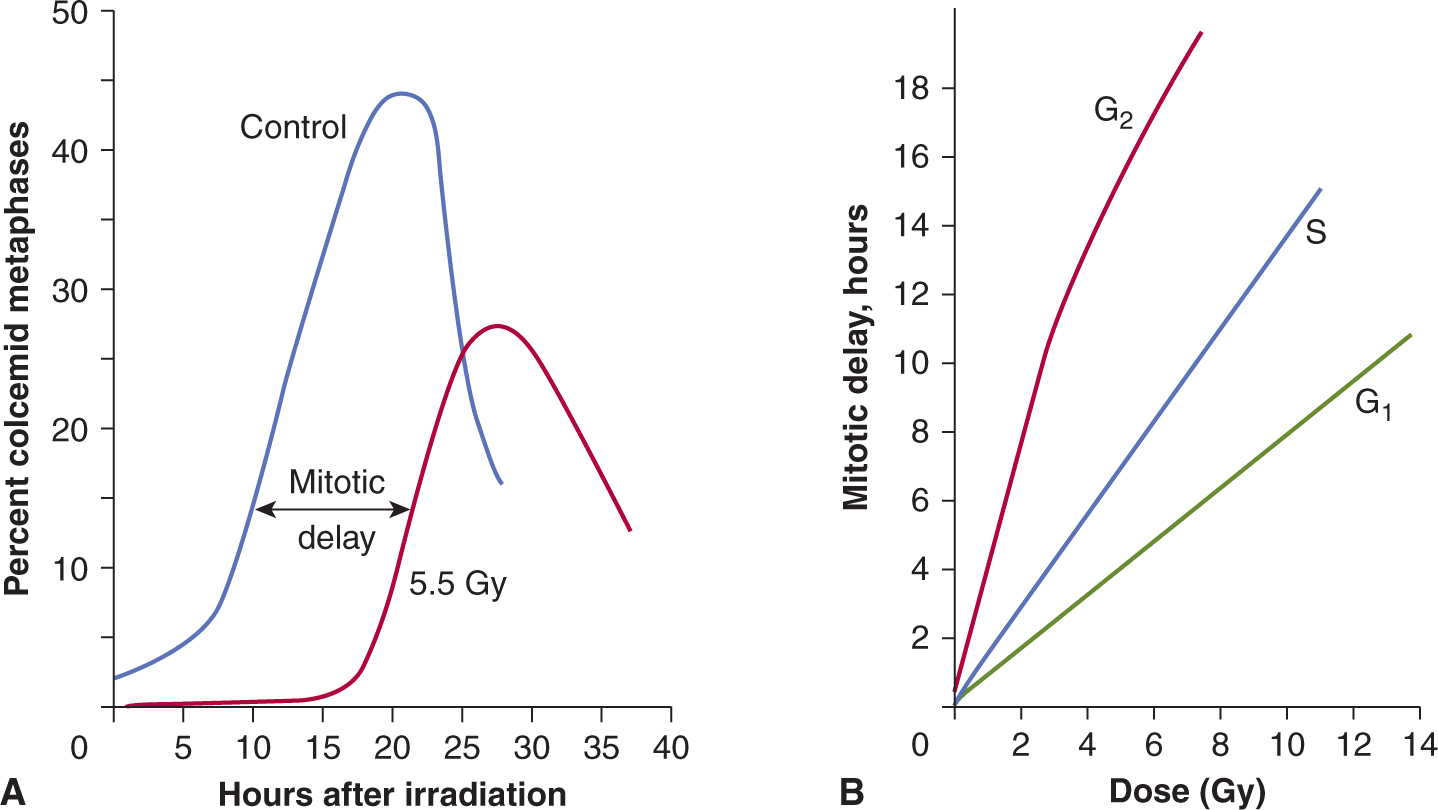
FIGURE 15–9 The effects of radiation on the progression of cells into mitosis after the treatment. A) At time zero, the cells are placed in medium containing colcemid, a drug that arrests cells in mitosis, and the percentage of cells that accumulate in mitosis is plotted as a function of time. The decline in the curves at late times is a result of cells escaping the drug-induced block or dying. The mitotic delay caused by a radiation dose of 5.5 Gy displaces the curves for the radiation-treated cells to the right. B) Cells are irradiated when in different phases of the cell cycle and the mitotic delay observed is plotted as a function of radiation dose. (Whitmore GF, Till JE & Gulyas S, unpublished data.)
Cells in different phases of the cell cycle have different radio-sensitivity (Terasima and Tolmach, 1961; Wilson, 2004). This is illustrated for Chinese hamster cells by the survival curves shown in Figure 15–10A. If a single radiation dose is given to cells in different phases (ie, a vertical cut is taken through the curves in Fig. 15–10A), then a pattern of cell survival as a function of cell-cycle position is obtained (Fig. 15–10B). Figure 15–10 shows that Chinese hamster cells in late S phase have the highest probability of survival after radiation (ie, are the most resistant), and that cells in mitosis (M phase) are the most sensitive. Although many cell lines appear to have a resistant period in S phase following irradiation in vitro, cell lines have variability in sensitivity just before mitosis in the G2 phase of the cell cycle (Wilson, 2004). For example, some oncogene-transfected cells show increased resistance in the G2 phase, whereas other cells, including DNA repair-deficient cells, show similar sensitivity throughout all phases of the cell cycle (Kao et al, 2001).
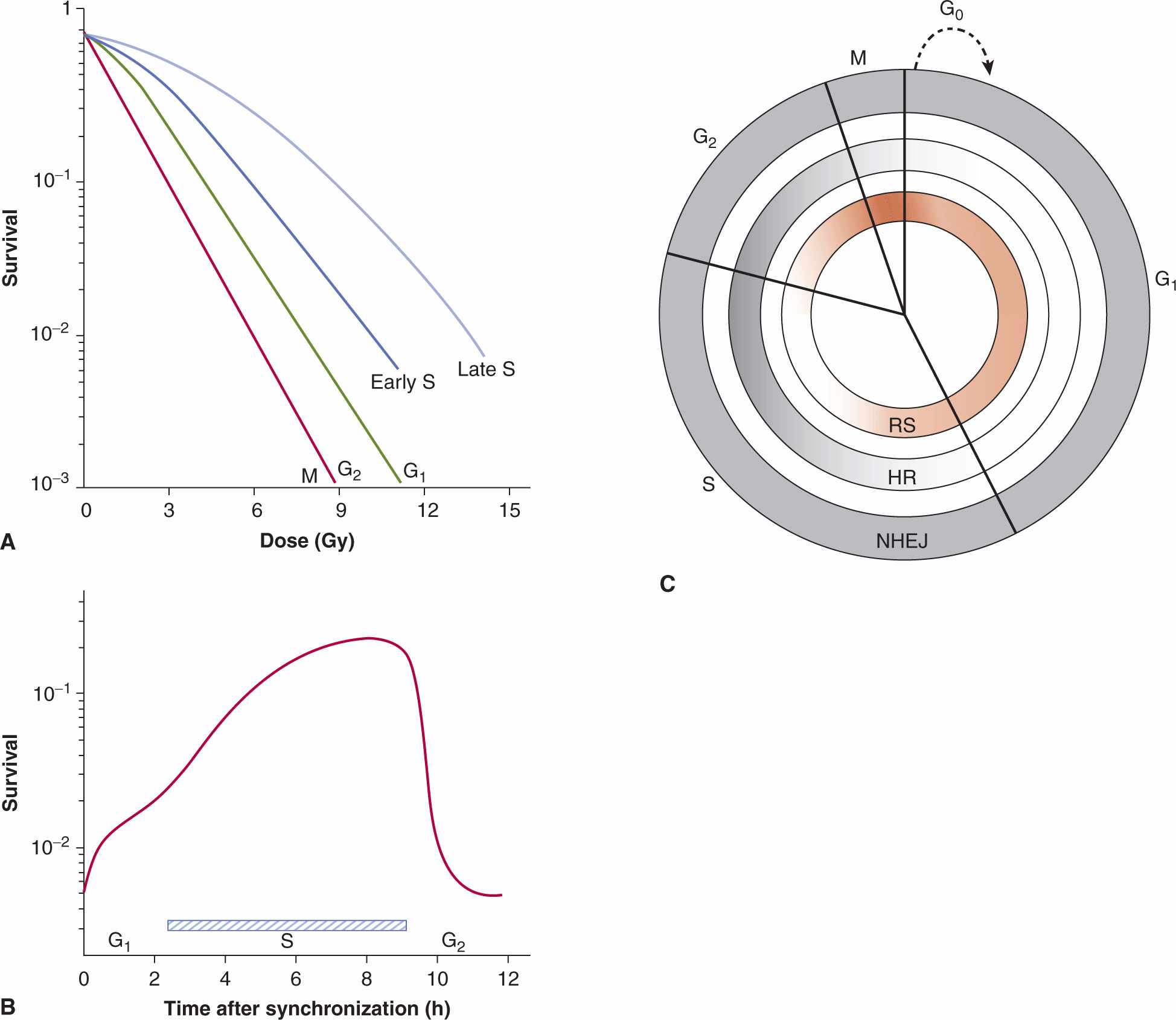
FIGURE 15–10 The effect of position in the cell cycle on cellular radiosensitivity. A) Survival curves for Chinese hamster cells irradiated in different phases of the cell cycle. B) Cells were selected in mitosis and irradiated with a fixed dose as a function of time of incubation after synchronization. The pattern of cell survival reflects the changing cellular sensitivity as the cells move through the cell cycle. C) Diagram indicating the active repair mechanisms during the various cell cycle phases and relative radiosensitivity. HR, Homologous recombination which occurs during the S and G2 phases of the cell cycle; NHEJ, nonhomologous end-joining recombination that occurs in all phases of the cell cycle; RS, relative radiosensitivity. Dark shading indicates activity of the particular repair pathway. Dark red shading indicates most radiosensitive portions of the cell cycle. (From Rothkamm et al, 2003.)
The molecular biology of the mammalian cell cycle and its response to DNA damage (including that of ionizing radiation) is discussed in detail in Chapter 5, Section 5.4 and Chapter 9, Section 9.3. The ATM (ataxia-telangiectasia mutated) protein plays a role in initiating checkpoint pathways in all 3 cell-cycle phases (Shiloh, 2003). G1 cell-cycle arrest following irradiation centers around an intact ATM-p53/CDC25A-RB pathway and decreased activity of CYCLIN D and E complexes. This leads to continued hypophosphorylation of the retinoblastoma (RB) protein at the G1–S interface and blocking of the initiation of DNA replication. Consequently, radiation-induced G1 arrest is abrogated in cells that lack functional p53, ATM, or RB proteins (Fei and El-Deiry, 2003; Cuddihy and Bristow, 2004). Although somewhat controversial, it is likely that loss of p53 protein function (and an abrogated G1 checkpoint) do not lead to radiosensitivity or radioresistance in comparison with those cells having normal p53 protein function; in contrast, mutant p53 has been reported to confer a radiosensitive phenotype (Choudhury et al, 2006). The S-phase checkpoint is controlled though ATM-mediated phosphorylation of the BRCA1, NBS1, and FANCD2 proteins that modify the activity of transcription factors (ie, E2F) and DNA replication proteins (RPA [replication protein A], PCNA [proliferating cell nuclear antigen]) during S-phase and DNA replication (Iliakis et al, 2003; see Chap. 5, Sec. 5.4).
There are 2 “checkpoints” in irradiated G2 cells. The G2/M checkpoint occurs early after radiation, is transient, is ATM-dependent and dose-independent (between 1 and 10 Gy). This checkpoint controls the entry into mitosis of cells that were in G2-phase at the time of irradiation. The “G2 accumulation checkpoint” is independent of ATM, but dependent on dose and ensures that cells that pass through earlier cell-cycle phases with DNA damage do not enter mitosis (Xu et al, 2002) The G2 arrest following exposure to ionizing radiation probably allows damaged DNA to be repaired prior to mitosis since DNA repair activity has been detected during the radiation-induced G2 delay and has been related to cellular radiosensitivity (Nagasawa et al, 1994; Kao et al, 2001). Molecular alterations that have been associated with the onset and duration of the G2 delay following radiation treatment include (a) cytoplasmic sequestration or decreased expression and stability of the CYCLIN-B protein and (b) inhibitory phosphorylation of the p34CDC2 protein after inactivation of the CDC25C phosphatase following radiation-induced activation of CHK1 or CHK2. These effects result in prevention of the formation of nuclear cyclin-B-p34CDC2 complexes, which are required for G2 progression (see Chap. 9, Sec. 9.2.3). Cells lacking either ATM or ATR-CHK1 function exhibit a defective G2 check-point after irradiation (Xu et al, 2002 and Chap. 5, Sec. 5.4).
Tumor cells often exhibit an aberrant G1 cell-cycle checkpoint because of defects in the ATM-p53-RB pathways, while the G2 cell-cycle checkpoints remain intact. There have been attempts to develop drugs that abrogate the G2 checkpoint (eg, caffeine, methylxanthines, or 7-hydroxystaurosporine [UCN-01]) in tumor cells to potentiate the cytotoxicity of ionizing radiation over that of normal cells. These drugs lead to the induction of premature mitosis and mitotic catastrophe in the treated cells. For example, UCN-01 preferentially sensitizes p53-mutated, radioresistant tumor cells to ionizing radiation. Identification of the targets of caffeine and UCN-01 (ie, ATM/ATR and CHK1/2) has led to interest in the development of this class of agent for the radiosensitization of tumors (Tenzer and Pruschy, 2003; Choudhury et al, 2006; Mitchell et al, 2010).
15.4.2 Cellular and Molecular Repair
The repair of cellular damage between radiation doses is the major mechanism underlying the clinical observation that a larger total dose can be tolerated when the radiation dose is fractionated. The shoulder of the survival curve reflects accumulation of sublethal damage that can be repaired (Elkind and Sutton, 1960; Fig. 15–11). When Chinese hamster cells were incubated at 37°C (98.6°F) for 2.5 hours between the first and second radiation treatments, the original shoulder of the survival curve was partially regenerated, and it was completely regenerated when the cells were incubated for 23 hours between the treatments (Fig. 15–11A). When the interval between 2 fixed doses of radiation was varied (Fig. 15–11B), there was a rapid rise in survival as the interval was increased from zero (single dose) to about 2 hours. This was followed by a decrease before the survival rose again to a maximum level after about 12 hours. This pattern of recovery is a result of 2 processes. Repair of sublethal damage (SLDR) accounts for the early rise in survival. Because cells that survive radiation tend to be synchronized in the more resistant phases of the cell cycle, their subsequent progression (inevitably into more sensitive phases) leads to a reduction in survival at 4 hours. Continued repair and repopulation explain the increases in survival at later times (see Chap. 16, Sec. 16.6). This pattern of SLDR has been demonstrated for a wide range of cell lines.
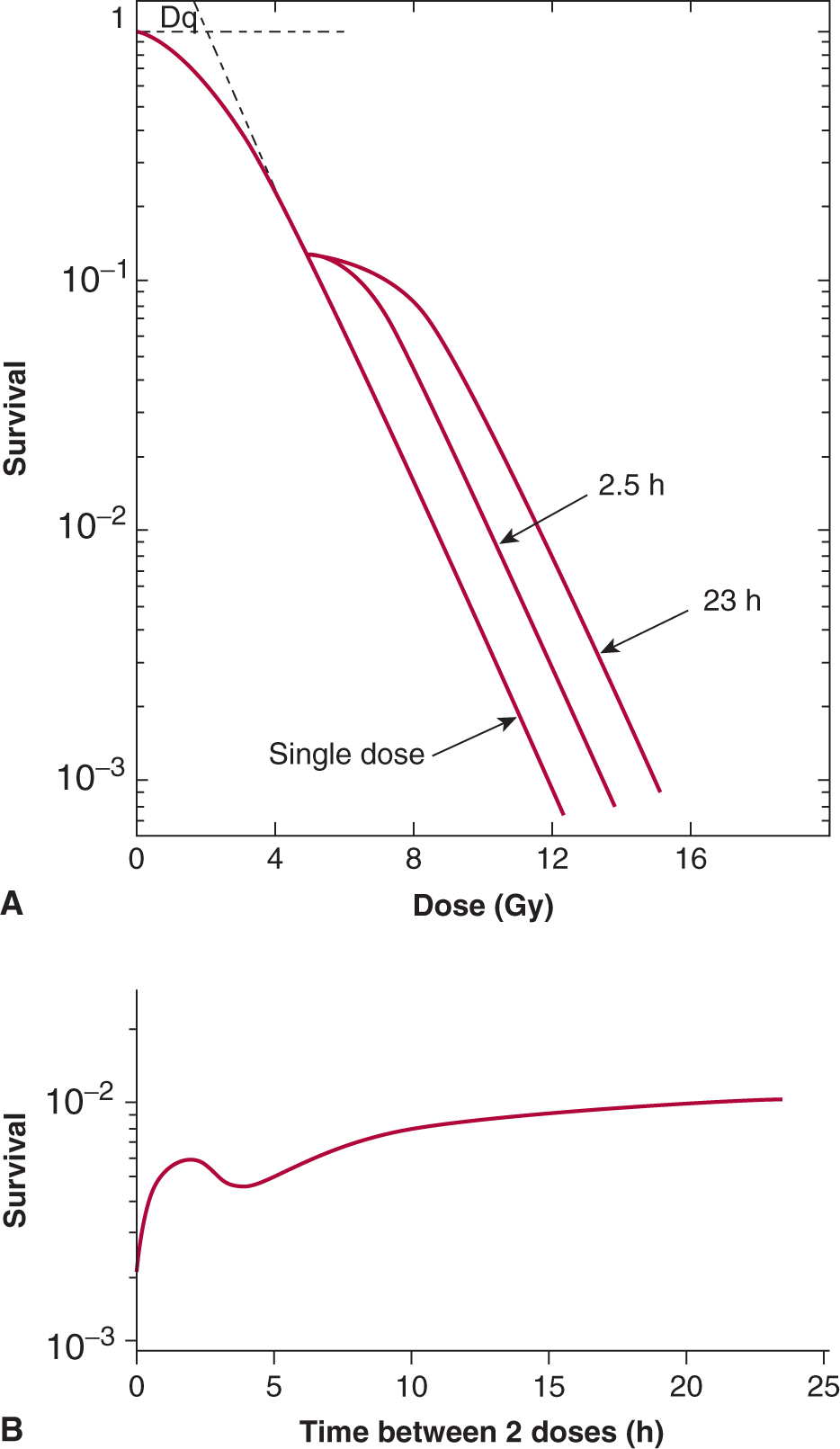
FIGURE 15–11 Illustration of the repair of sublethal damage that occurs between 2 radiation treatments. A) Survival curves for a single-dose treatment or for treatments involving a fixed first dose followed, after 2.5 or 23 hours of incubation (at 37°C), by a range of second doses. B) Pattern of survival observed when 2 fixed doses of irradiation are given with a varying time interval of incubation (at 37°C) between them. (Adapted from Elkind and Sutton, 1960.)
The repair capacity of the cells of many tissues in vivo has been demonstrated using cell survival and functional assays in vivo (Withers and Mason, 1974). An increase in total dose is required to give the same level of biological damage when a single dose (D1) is split into 2 doses (total dose D2) with a time interval between them (Fig. 15–12). The difference in dose (D2 – D1) is a measure of the repair by the cells in the tissue.
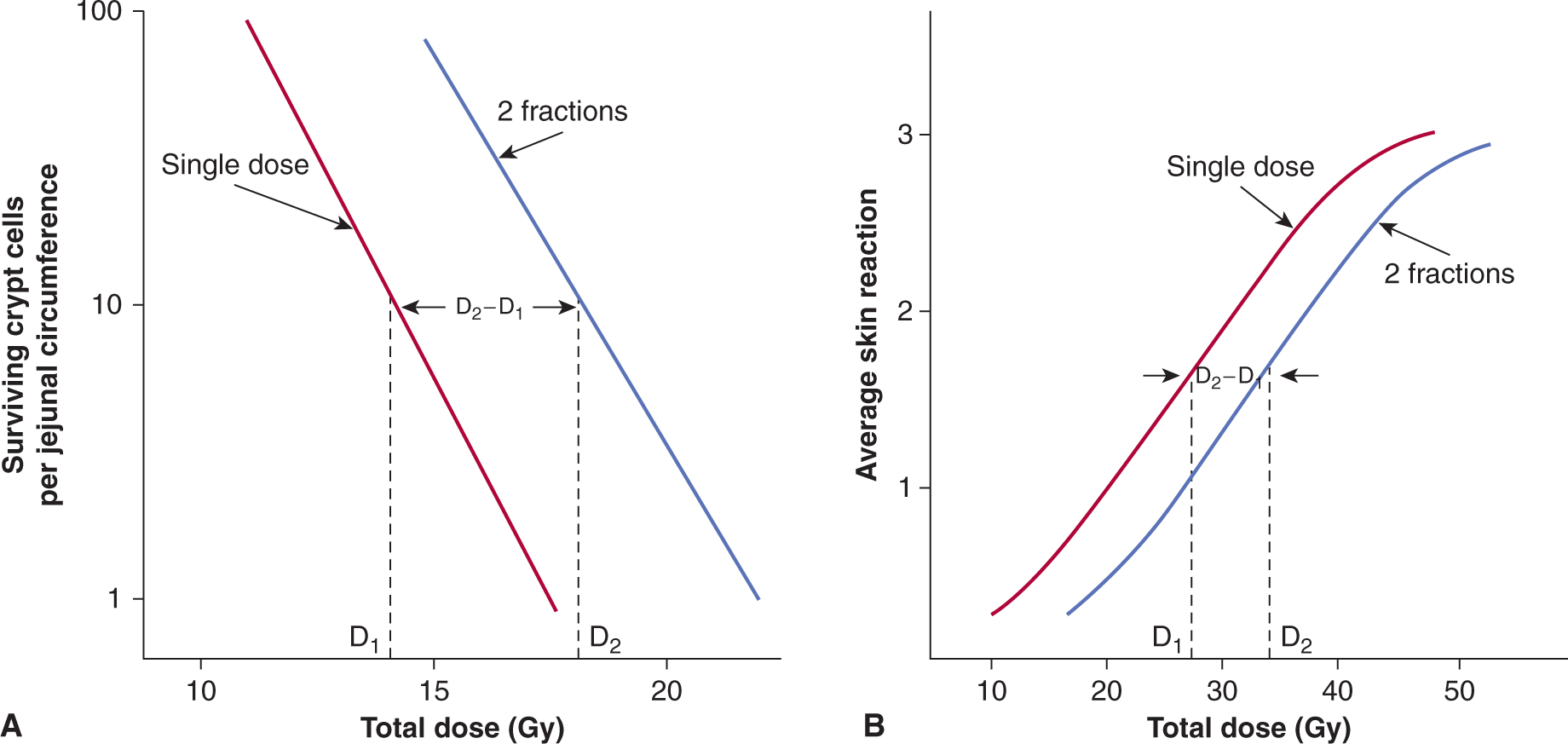
FIGURE 15–12 Repair of radiation damage in vivo. A) Survival curves for murine intestinal crypt cells γ-irradiated in situ with a single dose (red line) or with 2 equal fractions given 3 hours apart (blue line). (Modified from Withers and Mason, 1974.) B) Average skin reaction following x-irradiation of mouse skin with a single dose (red line) or 2 fractions given 24 hours apart (blue line). In both cases, an increase in total dose is required to give the same level of biological damage when a single dose (D1) is split into 2 doses (total dose D2) with a time interval between them. The difference in dose (D2 – D1) is a measure of the repair by the cells in the tissue.
The capacity of different cell populations to undergo SLDR is reflected by the width of the shoulder on their survival curve—that is, the Dq or D2 – D1 value. Survival curves for bone marrow cells have little to no shoulder, presumably because of their propensity to undergo radiation-induced apoptosis. Cells that demonstrate little or no evidence of cellular repair and that do not undergo radiation-induced apoptosis (such as fibroblasts derived from the radiosensitive disorders ataxia telangiectasia [AT] and Nijmegen breakage syndrome [NBS]; see Chap. 5, Sec. 5.5) also lack a shoulder to their survival curve and accumulate increased levels of DNA breaks (Fig. 15–13A) (Shiloh, 2003; Jeggo and Lavin, 2009). This relationship is shown in Figure 15–13 where the increased radiosensitivity is associated with an increased accumulation of residual and lethal DNA double strand breaks. Other cells (eg, jejunal crypt cells) can demonstrate a large SLDR for repair capacity (D2 – D1 value of 4 to 5 Gy – see Fig. 15–12A).
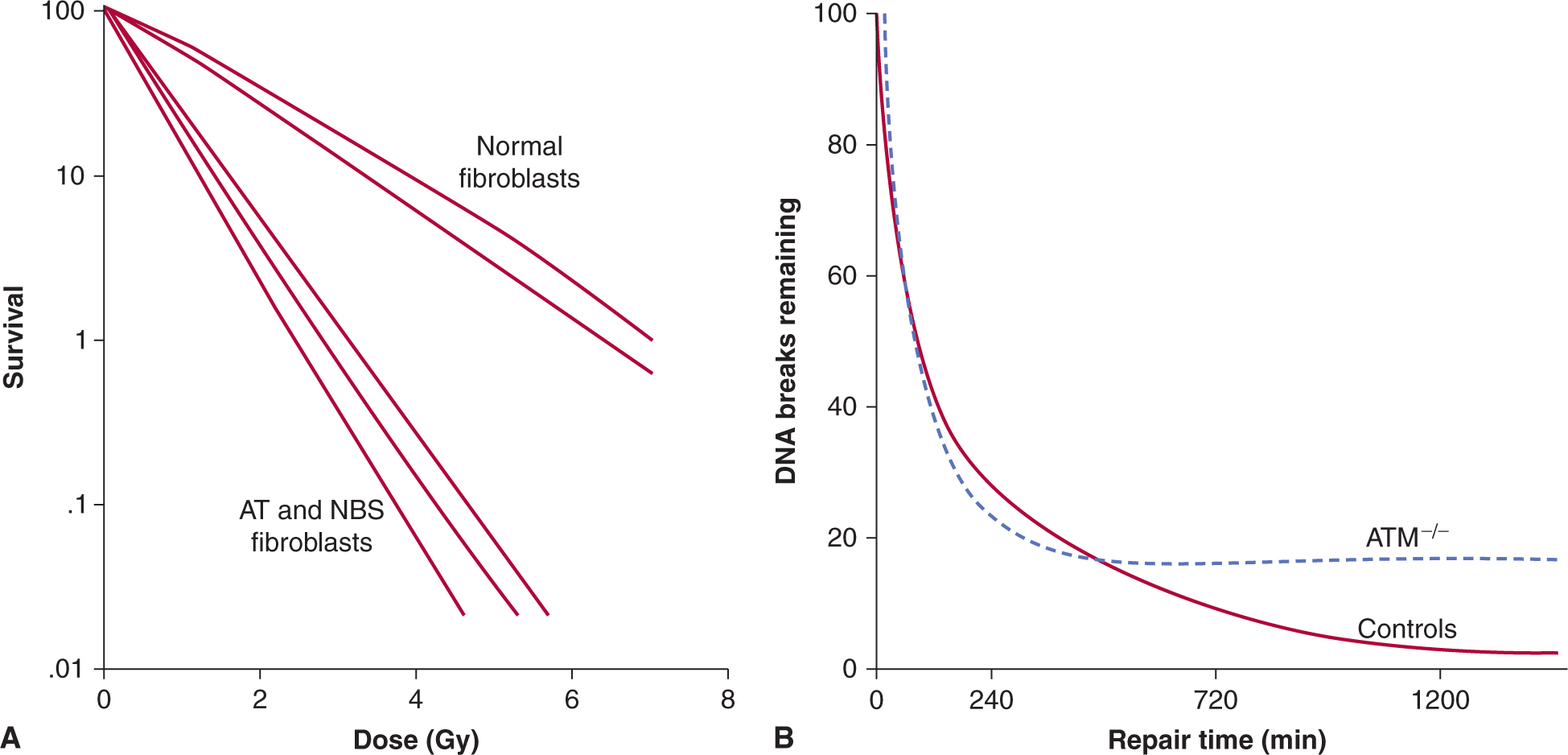
FIGURE 15–13 The relationship between radiosensitivity and DNA double strand breaks. In addition to aberrant cell-cycle checkpoints, increased radiosensitivity and subtle DNA double-strand break repair defects are associated with the AT and NBS disorders. Shown in (α) is the relative radiation survival for normal diploid fibroblasts versus that of AT or NBS fibroblasts. Note increased radiosensitivity following clinically relevant doses of 1 to 2 Gy. Shown in (B) are the DNA double-strand break rejoining curves (based on pulse-field gel electrophoresis) for AT cells relative to normal cells. The number of DNA double-strand breaks remaining is plotted against time following irradiation. Although the 2 sets of data initially have similar rates of DNA rejoining, the AT cells have increased numbers of residual DNA double-strand breaks relative to controls at later times postirradiation. (Modified from Girard et al, 2000.)
The effect of a given dose of radiation on human tissues and cells differs widely for exposures given over a short time (acute irradiation) and over an extended period of time (chronic irradiation given at a low-dose rate). Dose rates above approximately 1 Gy/min can be regarded as acute (single-dose) treatment and result in survival curves similar to that in Figure 15–7. As the total dose of X- or γ-rays is delivered at decreasing dose rates, the DNA damage in the cell diminishes progressively because of repair of the damage during the treatment. As a result, the shape of the radiation survival curve changes from one exhibiting a shoulder at high dose rates to one approaching linearity at low dose rates (Fig. 15–14A, B).
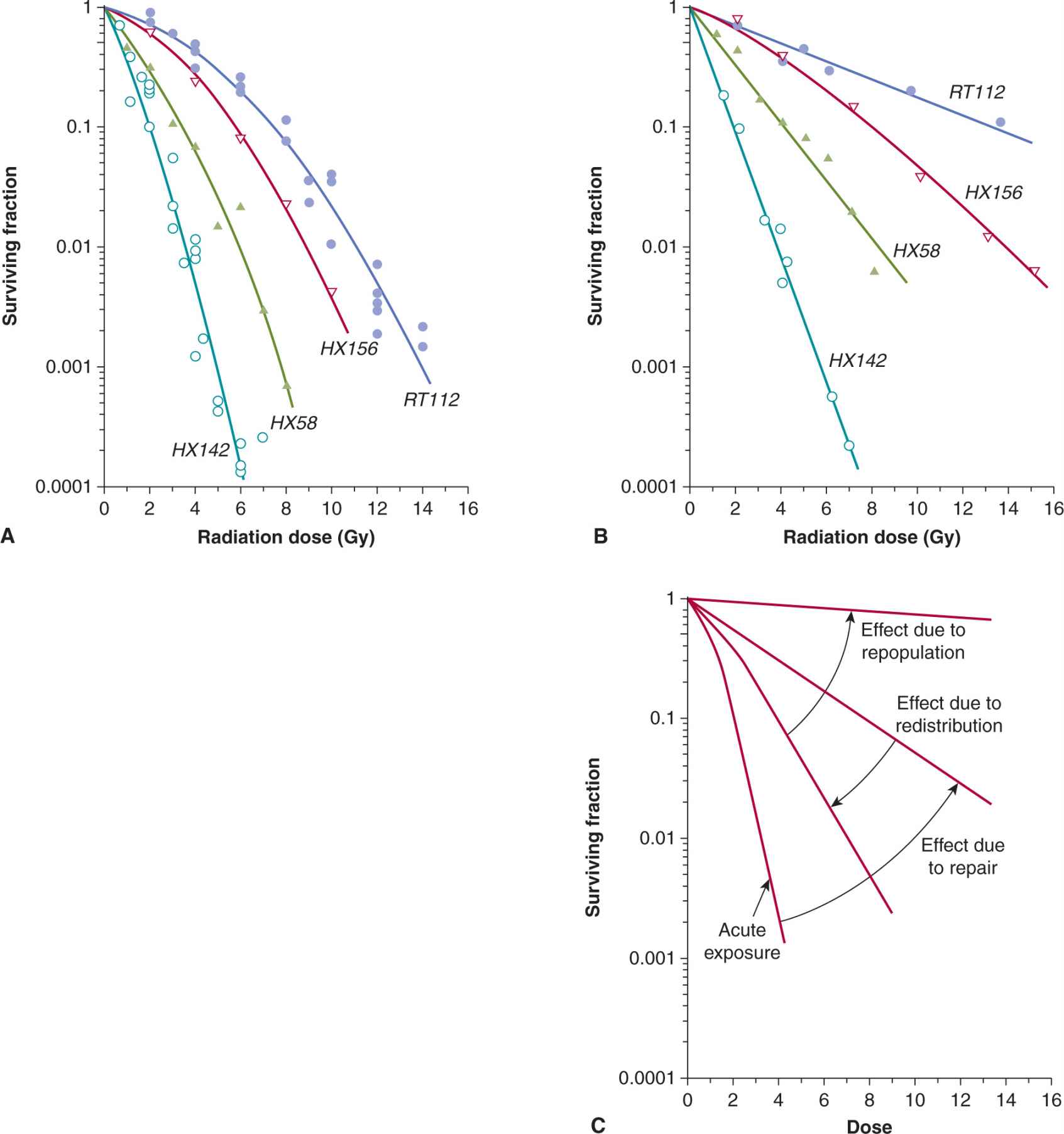
FIGURE 15–14 Survival curves for a series of human cancer cells lines irradiated under acute (high; >1 Gy/min) (A) or continuously (low; ~1Gy/h) (B) dose rates. (From Steel, 1991.) C) Schematic to illustrate the influence on the survival curve following continuous low-dose rate irradiation, of the processes of cellular repair, redistribution, and repopulation. (Data compiled by Dr. J.D. Chapman, Fox Chase Cancer Center, Philadelphia.)
The magnitude of the dose-sparing effect may be calculated as the relative survival under conditions of low-dose rate irradiation compared to survival under conditions of acute-dose rate irradiation. Cell lines with a greater capacity to repair sublethal damage will demonstrate a large dose-sparing effect relative to those cells that have limited capacity to repair the damage. In addition to cellular repair, low-dose rate irradiation can trigger the G1, S, and G2 checkpoints, slowing down the progression of the cells through the cycle. However, if the dose rate is low enough, the cells will continue to divide and repopulate. Because cells in late S phase are often the most radioresistant, they will preferentially survive, but eventually will move into the more sensitive phases of the cell cycle during radiation, a process termed redistribution. The process of repopulation leads to relative radioresistance of the cell population whereas cell-cycle redistribution leads to relative radio-sensitization. Most of the effect of cellular repair occurs in the range of dose rates of 1.0 to 0.01 Gy/min. Below approximately 0.1 Gy/min, the effects of cell-cycle progression (redistribution and the G2 block; see Sec. 15.4.1) become apparent; below approximately 0.01 Gy/min, the effects of cell repopulation will start to become evident as the radiation damage is not severe enough to trigger cell-cycle arrest (see Fig. 15–14C). Repair, repopulation, and redistribution are important for understanding dose fractionation in clinical radiotherapy, as described in Chapter 16, Section 16.6.
Cell survival can be increased by holding cells after irradiation under conditions of suboptimal growth, such as low temperature, nutrient deprivation, or high cell density. The latter conditions may reflect those experienced by G0-G1 populations of cells in growth-deprived regions of tumors (Malaise et al, 1989; see Chap. 12, Secs. 12.2 and 12.3). The property is a result of the repair of potentially lethal damage (PLDR), which usually results in a change in the slope of the cell-survival curve. Such repair may contribute to increased radiation survival observed in vivo for some transplantable cell lines when compared to the radiosensitivity of the same cells growing in vitro.
The molecular components of DNA repair pathway(s) are described in Chapter 5, Section 5.3. Data from a number of studies indicate that double-strand breaks (DSBs) are responsible for the majority of lethal damage induced by ionizing radiation (Jeggo and Lavin, 2009). The main pathways of repair of DNA-DSBs include homologous recombination (HR), which is maximally operational during S- and G2-phases, and nonhomologous end-joining (NHEJ), which is operational throughout the cell cycle (Rothkamm et al, 2003; Valerie and Povirk, 2003), as diagrammed in Figure 15–10.
There is no simple relationship between expression of DNA repair genes and relative radiosensitivity amongst normal or tumor cells that do not have a recognized genetic defect in DNA repair (Jeggo and Lavin, 2009). However, DNA repair capacity can influence cellular radiosensitivity, as indicated by the extreme radiosensitivity of cells from patients with DNA repair deficiency syndromes such as AT and the NBS (see Fig. 15–13 and Chap. 5, Sec. 5.5). Similarly, cells deficient in the BRCA1 or BRCA2 proteins, can have decreased HR-related repair and cell survival following radiation (Powell and Kachnic, 2003). A reduced capacity for repair of DNA DSBs is also observed among (radiosensitive) fibroblasts derived from severe combined immunodeficiency (SCID) mice in which deficient NHEJ is caused by a mutation in the enzyme DNA-PKcs, which is involved in recruiting repair proteins to the break site (see Chap. 5, Sec. 5.3.5). Indeed, mouse cells made deficient for NHEJ (ie, mouse knockouts for DNAPKcs or Ku70 genes) have exquisite radiosensitivity and defective rejoining of DNA-DSBs (see Fig. 15–15).
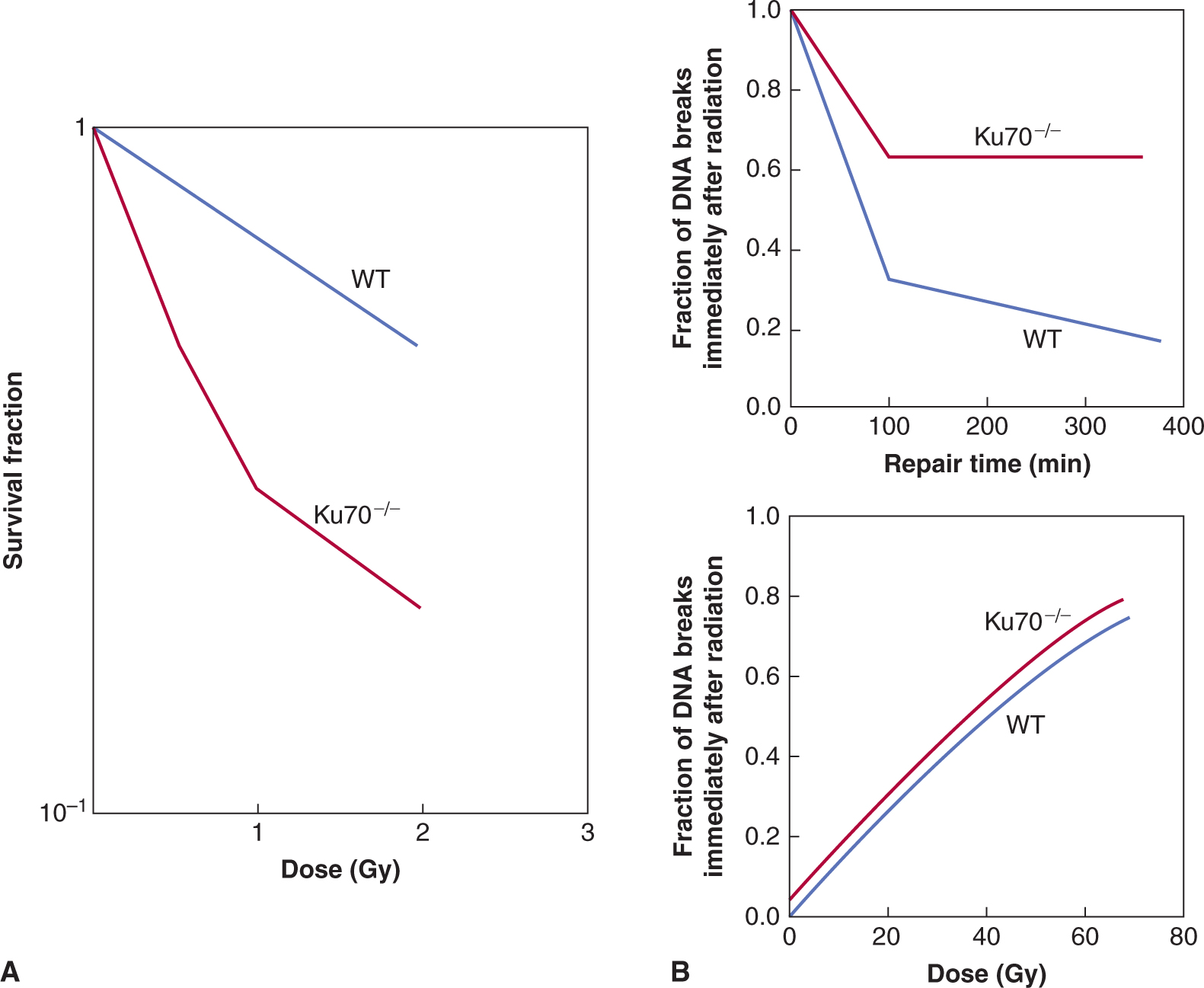
FIGURE 15–15 The role of NHEJ in DNA DSB repair and cellular radiosensitivity. A) The Ku70 protein, with the Ku80 and DNA-PKcs proteins, forms an important DNA-PK complex that initially catalyzes the repair of DNA-DSBs (see Chap. 5, Sec. 5.3.5 for details). As shown in (A), cells that are deficient in NHEJ proteins (eg, Ku70–/– fibroblasts) show exquisite radiosensitivity relative to normal wild-type (WT) cells. This is also true for Ku80- and DNA-PKcs-deficient cells. This increased radiosensitivity is a result of a reduced capacity for DNA break rejoining such that NHEJ-deficient cells have increased residual DNA breaks following irradiation leading to increased cell killing. This is illustrated in (B) (upper panel) where the number of remaining DNA DSBs at 400 minutes following irradiation is increased in Ku70–/– cells relative to WT cells. This is consistent with a DNA rejoining defect in the Ku70–/– cells. Note that the induction of DNA DSBs is similar between the 2 types of cells (lower panel), which, shows the number of DNA breaks induced for a given dose measured immediately following irradiation. (Modified from Ouyang et al, 1997.)
Broadly, cells can fall into 3 categories of sensitivity to ionizing radiation (Fig. 15–16A): Group I represents the “normal” case and Li-Fraumeni cells with p53-mutations and cells from patients with defects in DNA repair pathways not involved in DSB repair, such as nucleotide excision repair (NER), fall in this category. The G1/S checkpoint may be lost owing to p53-mutations, but this has no effect on the sensitivity of most cells to IR. Group II includes cells that have defects in HR or in “mediator” proteins, such as RNF168 (see Chap. 5, Sec. 5.5 and Table 5–1). Group III is the most radiosensitive and includes cells with defects in NHEJ and with mutations in the NBS1, ATM, or DNA ligase IV genes from DNA repair disorders (Girard et al, 2000). These cells are sensitive as a consequence of errors in end-processing and end-joining of DSB, defects in chromatin modifications that prevent efficient rejoining of DSBs, and inefficient ATM-mediated G2/M-checkpoints (Beucher et al, 2009) (see Chap. 5, Sec. 5.5 and Table 5–1).
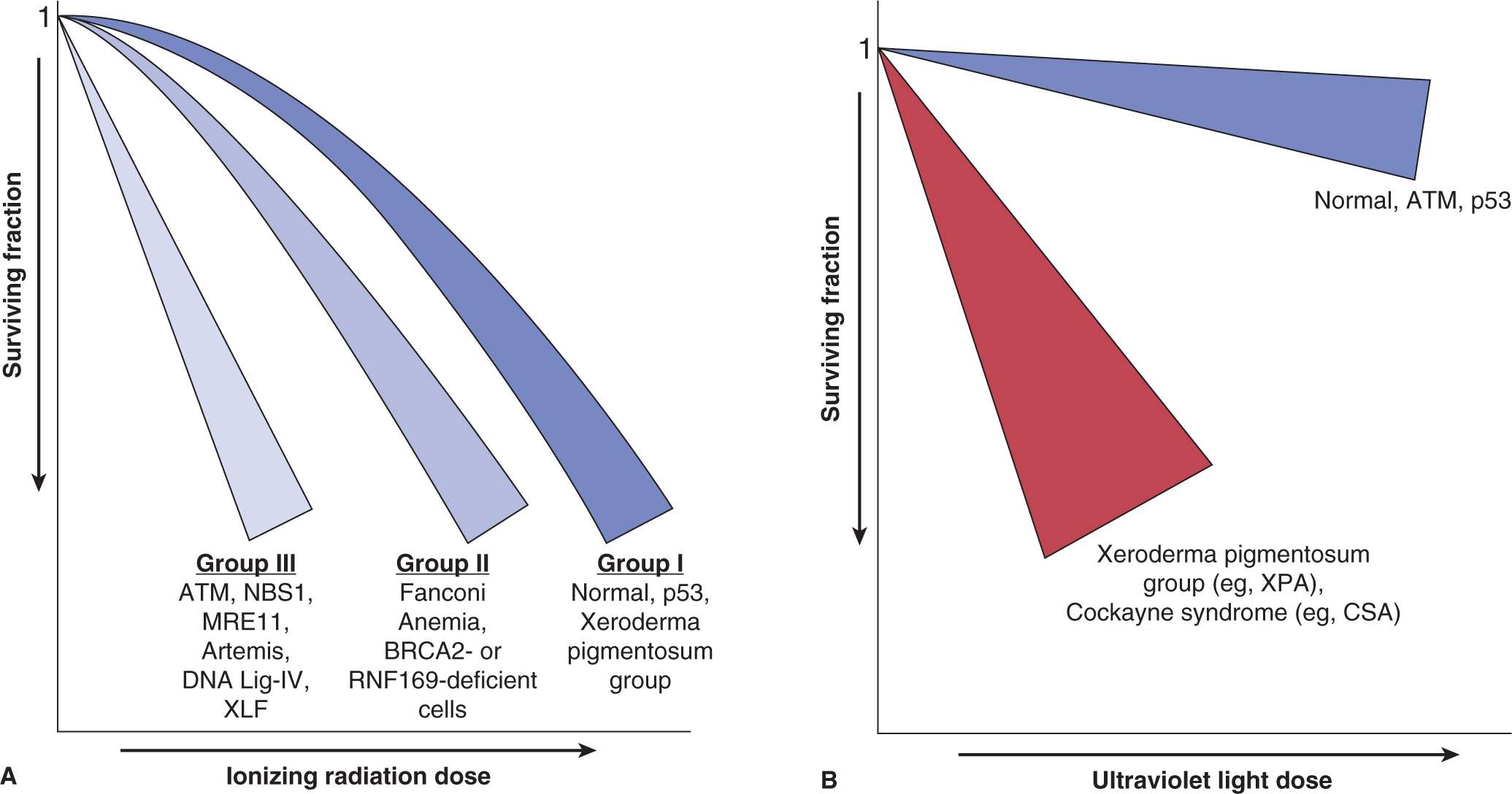
FIGURE 15–16 Schematic of clonogenic survival curves showing differences in survival following IR (A) and UV (B) radiation for cells deficient in genes involved in DNA repair or DNA damage signaling. These data are derived from experiments using DNA repair-deficient cells from DNA instability syndromes (details of these syndromes are as discussed in Chapter 5 [see Sec. 5.3.5]).
Most cells that show sensitivity to IR are not sensitive to UV radiation. Cells derived from people with Cockayne syndrome or xeroderma pigmentosum patients with NER defects are exquisitely sensitive to UV irradiation (Fig. 15–16B). This is consistent with the different types of damage caused by IR versus UV radiation and the different DNA repair pathways that are used (eg, DNA DSBs and single-strand breaks, repaired by HR and NHEJ versus cyclobutane pyrimidine dimers (CPDs) and 6-4PPs repaired by NER, respectively (see Chap. 5, Sec. 5.3 for details). It also highlights the repair of DSBs as the primary determinant of survival following IR.
Greater understanding of the relationship between deficient DNA repair and radiosensitivity has led to strategies designed to radiosensitize tumor cells. In human fibroblasts, small silencing RNAs (siRNAs; see Chap. 2, Sec. 2.4.3) or small molecule inhibitors have been used to decrease expression of endogenous DNA-PKcs or ATM, which results in defective DSB repair and an increase in residual (unrejoined) DNA DSBs, which leads to increased radiation cell killing (Peng et al, 2002; Thoms and Bristow, 2010). Similarly antisense RNA or pharmacological approaches (ie, the drug imatinib, which inhibits the interaction between the protein product of the c-abl oncogene and the DNA repair protein RAD51; see Chap. 17, Sec. 17.3.1) have been used to decrease expression of DNA repair proteins with resultant radiosensitization (Collis et al, 2001). Inhibiting the repair of DNA base damage and single-strand DNA breaks with inhibitors of poly (ADP-ribose) polymerase (PARP; see Chap. 17; Sec. 17.3.2) can also lead to radiosensitization (Chalmers et al, 2010). However, the degree of radiosensitization and DSB repair may differ in vitro and in vivo because of the additional effects of the microenvironment, which may lead to differential DSB induction and altered expression and function of DSB repair pathways (Chan et al, 2009; Jamal et al, 2010).
There may be a therapeutic advantage to targeting DNA repair in combination with radiotherapy in that cell kill in tumor cells can be increased relative to cell kill in normal tissues. In some tumor cells, DNA repair pathways may be nonfunctional so that the tumor cells are radiosensitized if there are tumor-specific defects in HR or NHEJ. Furthermore, some drugs, such as PARP inhibitors, may be selectively toxic to HR-defective tumor cells based on synthetic lethality, which could be used to decrease the number of tumor clonogens prior to or during radiotherapy (Thoms and Bristow, 2010).
15.4.3 Intracellular Signaling, Gene Expression, and Radiosensitivity
Intrinsic changes in gene expression in tumors can influence response to radiation, and irradiation can modify the expression of some genes. Biochemical processes in cells, such as DNA, RNA, or protein synthesis, respiration, or other metabolism can be inhibited by irradiation, but this usually requires quite large doses of the order of 10 to 100 Gy. Clinical doses of radiation can affect the expression of a number of genes involved in the response of cells to stress and this may change their properties. Aberrant expression of oncogenes or tumor-suppressor genes may increase the intrinsic cellular radioresistance of human and rodent cells (Haffty and Glazer, 2003). For example, increased radiation survival has been observed in selected cell lines following the transfection of a single oncogene, such as activated Ras, Src, or Raf (Kasid et al, 1996; Gupta et al, 2001). This has led to studies designed to radiosensitize tumor cells by the inhibition of oncogene function using inhibitors of intracellular signaling pathways or antisense RNA to decrease oncogene overexpression (Kasid and Dritschilo, 2003). Figure 15–17 indicates pathways that may be targeted and the preclinical and clinical data to support such targeting in tumor cell radiosensitization strategies (Begg et al, 2011)
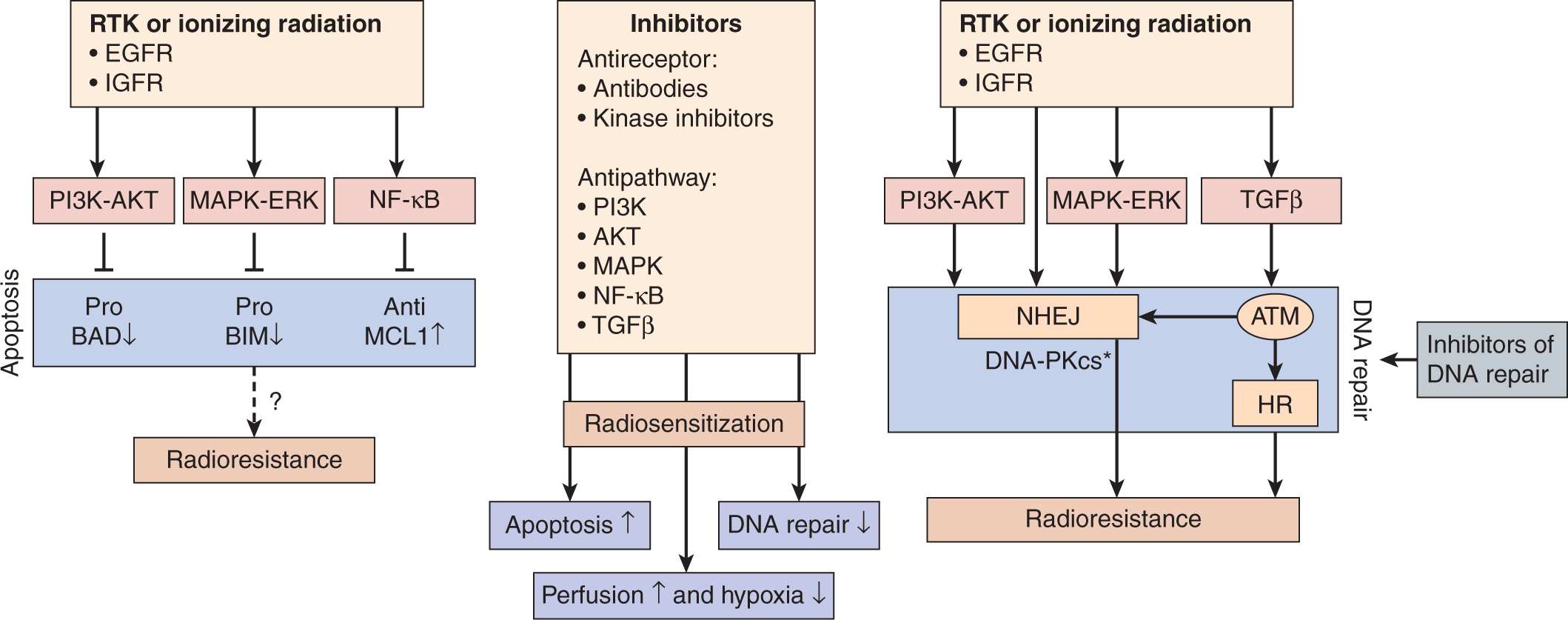
FIGURE 15–17 Targeting signal transduction. Growth factor receptor activation by mutation or overexpression, or mutations in oncogenes (such as RAS) or tumor suppressor genes (such as PTEN) can lead to signaling through the phophatidylinositol-3 kinase (PI3K)-AKT, mitogen-activated protein kinase (MAPK)-extracellular signal regulated kinase (ERK), nuclear factor-κB (NF-κB), and transforming growth factor-β (TGFβ) pathways. Such signaling can affect radiosensitivity by decreasing apoptosis (left) or increasing DNA repair (right). AKT, MAPK, and NF-κB signaling can all lead to phosphorylation and inactivation of proapoptotic proteins or activation of antiapoptotic proteins. Altering apoptosis does not always lead to changes in clonogenic cell survival (dashed arrow with question mark). Activation of the AKT and MAPK pathways leads to the activation of the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), a central protein in DSB repair by NHEJ. Direct inhibition of DNA repair may also lead to radiosensitization by targeting the ATM and DNA-PKcs kinases or PARP proteins. DNA-PKcs can also be activated by the receptor tyrosine kinase (RTK) epidermal growth factor receptor (EGFR) after it is translocated to the nucleus. There is a strong correlation between DNA repair capacity, particularly for DSBs, and radiosensitivity. Ionizing radiation can also activate the PI3K, MAPK, and NF-κB pathways. Inhibition of signaling pathways following irradiation can therefore reverse (Begg et al, 2011) tumor cell radioresistance. Some inhibitors have also been shown to affect tumor vasculature, leading to improved perfusion and reduced hypoxia (see also Chap. 16, Sec. 16.2.5). Asterisk indicates activation. (Taken from Begg et al, 2011.)
When the Ras oncogene undergoes mutation, it is permanently activated in the guanosine triphosphate (GTP)-bound signaling state, providing proliferative signals in the absence of growth factor ligands, leading to altered cell growth, transformation, and, occasionally, radioresistance (see Chap. 8, Sec. 8.2.3). However, increased radioresistance is more commonly observed in cells transfected with an activated Ras gene in combination with a nuclear cooperating oncogene, such as c-Myc or mutant p53 (McKenna et al, 1990; Bristow et al, 1996). Downstream to RAS, the RAS-MEK-ERK and phosphatidylinositol-3 kinase (PI3K)-AKT/PKB pathways (see Chap. 8, Sec. 8.2.5) have been linked to tumor radioresistance (Bussink et al, 2008). Using antisense oligonucleotides or silencing RNA against human Raf leads to increased cancer cell radiosensitivity (Woods Ignatoski et al, 2008; Kidd et al, 2010). RAS-mediated radioresistance in rat cells appears to be dependent on PI3K and RAF signaling pathways, and less on the MEK signaling pathway (Affolter et al, 2012). Inhibitors of RAS and PI3K signaling, such as LY294002 and wortmannin, significantly enhanced the response to radiation in lung, bladder, colon, breast, prostate, head and neck squamous cell carcinoma (HNSCC), and cervical cancer cells (Xiao et al, 2010). Although inhibitors of RAS protein prenylation or function (farnesyl transferase inhibitors) have been reported to enhance radiation-induced cytotoxicity among preclinical models of human breast, lung, colon, and bladder cancer cells expressing mutated H- or K-ras genes, the use of these agents in the clinic has been limited by the lack of biomarkers that reflect a precise drug targeting of RAS versus other intercommunicating signaling pathways (Rengan et al, 2008).
Activation of the PI3K-AKT/PKB pathway is associated with 3 major mechanisms of tumor radioresistance: intrinsic radio-resistance, tumor-cell proliferation, and hypoxia. Activation of this pathway can be caused by stimulation of receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR; see Chap. 8, Sec. 8.2.1). In clinical trials, an independent association has been noted between expression of activated AKT and outcome of treatment of head and neck cancer with radiotherapy (Bussink et al, 2008). More recent data implicate the Akt gene in DSB repair, as AKT can stimulate the accumulation of DNAPKcs at DNA-DSBs and promote DNA-PKcs activity during NHEJ, and itself can bind to DSBs in vitro following irradiation (Fraser et al, 2011; Toulany et al, 2012). Consequently, there is interest in the use of AKT inhibitors as clinical radiosensitizers given the overexpression of activated AKT in many tumors.
Tyrosine kinase activity of EGFR is increased following cellular exposure to radiation and addition of exogenous epidermal growth factor (EGF) to cell culture renders cells relatively radio-resistant. Both EGFR and the related HER-2/neu receptor are overexpressed in a wide variety of epithelial tumors (head and neck squamous cell cancers (HNSCC), gliomas, breast, lung, colorectal and prostate cancers) and this overexpression has been associated with poor clinical outcome following radiotherapy due in part to increased tumor cell repopulation during therapy (Verheij et al, 2010). Targeting EGFR or human epidermal growth receptor 2 (HER-2)/neu signaling using monoclonal antibodies or specific inhibitors (eg, cetuximab-EGFR or trastuzumab-HER-2) leads to radiosensitization, in vitro and in vivo. These drugs are being tested in combination with radiotherapy and chemotherapy in randomized, multicenter clinical trials (Verheij et al, 2010; see Chap. 16, Sec. 16.2.5). Nuclear EGFR is now thought to also be part of the DNA-damage repair complex that interacts with proteins involved with the NHEJ repair pathway during ATM activation and chromatin relaxation during the sensing and repair of DSBs. Thus, inhibition of EGFR signaling during radiotherapy may lead to both decreased tumor cell repopulation and increased cell kill as a consequence of defective DSB repair (Dittmann et al, 2011; Yu et al, 2012).
The radiosensitivity of cells may be influenced by the addition of exogenous growth factors or hormones in receptor-positive cells before or after irradiation. The insulin-like growth factor-1 receptor (IGF-1R) is a cell-surface receptor with tyrosine kinase activity that has been linked to increased radioresistance. IGF-1R is expressed at low levels in AT cells; this may, in part, contribute to their radiosensitivity, as reintroduction of IGF-1R, or addition of exogenous insulin-like growth factor (IGF), can increase their radioresistance (Peretz et al, 2001). Other data implicate a role for IGF-1R directly in DSB repair, at least in part via the HR pathway and small molecule IGF-1R kinase inhibitors can decrease radioresistance in tumor cells (Turney et al, 2012).
15.4.4 Radiation-Induced Changes in Gene Expression
Irradiation can modify intracellular signaling through modification of the activity of tyrosine kinases, mitogen-activated protein (MAP)-kinases, stress-activated protein (SAP)-kinases, and RAS-associated proteins (Ruiter et al, 1999; Dent et al, 2003; Schmidt-Ullrich et al, 2003). Tyrosine phosphorylation is involved in several DNA damage response pathways; an example is activation of the c-ABL pathway, which phosphorylates RAD51, a DNA repair protein at sites of DNA damage. Genes induced by radiation include those encoding cell-cycle-related proteins (eg, growth arrest after DNA damage [GADD] genes, p34CDC2, CYCLIN B, p53), growth factors, and cytokines (eg, platelet-derived growth factor [PDGF], transforming growth factor beta [TGF-β], bFGF, tumor necrosis factor alpha [TNF-α]), and enzymes (eg, plasminogen activator). Liberation of inflammatory cytokines such as TNF-α and interleukin-1 (IL-1) by cells following radiation damage may lead to a continuing cascade of cytokine production, which may be responsible for the acute inflammation and late-onset fibrosis observed in some irradiated tissues (see Chap. 16, Sec. 16.5.3).
Induction of the expression of early response genes (ie, within seconds to minutes) by irradiation can be initiated by damage to the plasma membrane or to nuclear DNA (Criswell et al, 2003). Some early response genes, such as the early growth response factor (EGR-1) and p21 Cdk-inhibitor proteins (see Chap. 9, Secs. 9.2 and 9.3), contain radiation-responsive regulatory domains in their promoter regions, which can facilitate their rapid induction by radiation (Hallahan et al, 1995). These sequences might be used in radiation-induced gene therapy as vectors to drive expression of suicide genes (eg, TNF-α) for tumor therapy. Synthetic enhancers of gene expression designed for use with radiation utilize short motifs of sequence CC(A/T)6GG (ie, radiation-responsive elements) derived from the EGR-1 gene (Datta et al, 1992; Marples et al, 2002). Such constructs can be responsive to radiation at doses of 1 to 5 Gy. These tumor-targeting vectors might be used in clinical situations where the radiation volume can be tightly controlled to spare normal tissues using conformal radiotherapy (see Chap. 16, Sec. 16.2.2) and have shown promise in animal models (Mauceri et al, 2009).
Approaches using complementary DNA (cDNA) microarrays (see Chap. 2, Sec. 2.2.12) have led to the discovery that radiation-induced gene expression can be cell-type specific and while induction of some genes is dose-dependent across a range of doses, others are activated specifically at either low (ie, 1 to 3 Gy) or high doses (ie, 10 Gy) (Khodarev et al, 2001; Nuyten and van de Vijver, 2008; Rashi-Elkeles et al, 2011). Furthermore, gene expression following a given radiation dose can be substantially higher when solid tumors are irradiated in vivo than when the same cell line is irradiated in culture. However, studies of biopsies from human tumors have demonstrated that differences in radiation-induced gene expression are greater between patients’ tumors than within the tumor of a given patient (Hartmann et al, 2002). This observation supports the concept that “molecular profiling” of the tumors and normal tissues in individual patients may be able to predict radiation response.
MicroRNAs (miRNAs) are emerging as a class of endogenous gene modulators that control protein levels, thereby adding a new layer of regulation to the DNA damage response (see Chap. 2, Sec. 2.4.3). There is increasing interest in an association between miRNA expression in tumors and chemo- and radiosensitivity, both with regards to predicting and modulating sensitivity. These include the miRNAs of the Let-7 family, miR-21 and miR-200b, as inhibition of these miRNAs leads to increased radiosensitivity (Hu and Gatti, 2010).
The translation of preclinical to clinical testing for molecularly-based compounds is limited by multiple factors including the appropriate scheduling and toxicity of the agent, the interpatient and intratumoral variability of the expression of the molecular target and its role as a predictor of treatment response, and molecular crosstalk among redundant, parallel intracellular signaling pathways (Haffty and Glazer, 2003). Some of these limitations might be bypassed by simultaneous determination of multiple pathways using genomic and proteomic analyses (see Chap. 2) of both normal and tumor tissues as the basis for selection of the best molecular agents to be combined with radiation treatments (Ma et al, 2003; Ishkanian et al, 2010; Begg, 2012). The majority of proteins involved in DNA repair undergo posttranslational modification or novel protein–protein interactions following irradiation at sites of DNA damage. Because such modifications would not be detected using cDNA microarray analyses, which detect alteration of messenger RNA expression rather than altered protein levels, proteomic anaysis (see Chap. 2, Sec. 2.5) of tissue samples or sera may be required to determine molecular pathways that lead to radio-response in patients (Bentzen et al, 2008).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


